Reakcja eliminacyjna – Wikipedia
W chemii organicznej, a eliminacja (Lub β-élimination ) jest reakcją organiczną, która przekształca podstawioną alcanię (halogenoalkany, alkohole itp.) w pochodną etylenową, nawet alceen, jeśli cząsteczka początkowa, oprócz grupy początkowej, jest tylko łańcuchem węglowym typu ALCA.
Warunki są oprócz twardych warunków podstawienia nukleofilowego, ścisła reakcja wielu aspektów i konkurencyjna: eliminacja występuje w obecności silnej zasady i przez ogrzewanie mieszaniny reakcyjnej.
Jednym z głównych przykładów eliminacji jest transformacja halogenoalcanu w alcene; Następnie mówimy o dehydrohalogenacji:
Najczęściej stosowanymi zasadami są wodorotlenek sodu (na + , Do – ), wodorotlenek potasu (k + , Do – ), nawet jony alkoholowe (RO – ) (uzyskane przez reakcję sodu w ROH, bezwodnym alkoholu).
Podobnie jak w przypadku podstawienia nukleofilowego, występuje monomolekularna eliminacja i eliminacja bimolekularna, które są powszechnie odnotowane odpowiednio „E1” i „E2”.
W pozostałej części tego artykułu przykłady podawane są z halogenizowaną pochodną jako odczynnik.
Kinetyka E1 [[[ modyfikator |. Modyfikator i kod ]
Monomolekularna reakcja eliminacji odbywa się w dwóch etapach: monomolekularny etap, który obejmuje jedynie halogeniczną cząsteczkę pochodną z boku odczynników, która jest etapem kinowym (lub Ograniczenie ), a następnie krok bimolekularny. W konsekwencji kinetyka reakcji, przy aproksymacji determinacji kinetycznie, zależy tylko od stężenia gatunku RX.
Reakcja charakteryzuje się globalną kinetyką 1, częściowych rzędów 1 w odniesieniu do halogenowanej pochodnej i 0 w porównaniu z podstawą B | :: W = k [Rx], gdzie:
- [Rx] reprezentuje stężenie halogenizowanej pochodnej w mol/L;
- W reprezentuje prędkość reakcji eliminacji. Jest wyrażany w mol/s;
- k jest stałą prędkości: w rzeczywistości jest to krok kinetycznie ograniczający ( k Pierwszy ). Jego jednostka, L/S, odstąwa od tych z dwóch innych terminów.
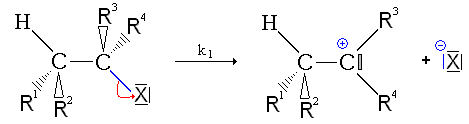
Mechanizm E1 [[[ modyfikator |. Modyfikator i kod ]
Reakcja odbywa się w dwóch etapach:
- Pierwszy krok: odejście z nukleoffuge, tworzenie karbokacji. Ten etap tworzenia karbokacji jest powolnym, odwracalnym krokiem i narzuca jego prędkość na całą reakcję;

Stereoselektywność i regioselektywność [[[ modyfikator |. Modyfikator i kod ]
Stereochemia [[[ modyfikator |. Modyfikator i kod ]
Jak pokazano w mechanizmie, przejście przez karbokacja, która jest płaską strukturą, usuwa każdy element stereospecyficzności w przeciwieństwie do (patrz kontynuacja) do mechanizmu eliminacji bimolekularnej.
Ale jeśli mogą powstać dwa geometryczne stereoizomery, nie tworzą się w tych samych proporcjach. Najbardziej stabilny izomer, to znaczy ten, którego energia jest najniższa, jest głównie utworzona (jest to zasada Zaïtseva). Dlatego jest to izomer E, który powstaje głównie przy braku mezomerii. Z drugiej strony, jeśli podwójne wiązanie jednego z dwóch produktów jest przenoszone przez mezomerię, to właśnie zostanie utworzone głównie. Następnie mówimy o częściowej stereoselektywności.
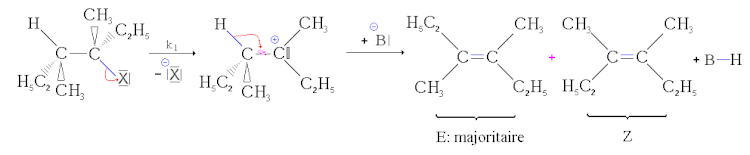
Regiochemia [[[ modyfikator |. Modyfikator i kod ]
Kiedy może powstać kilka izomerów konstytucyjnych, jest on preferencyjnie powstały przez najbardziej stabilny termodynamicznie alceen. Jeśli istnieją tylko grupy alkilowe, jest to najbardziej podstawiony alceen. Jeśli mezomerie mogą interweniować, jest to najbardziej skoniugowana alcena, która jest większością.
Następnie mówimy o regioselektywności lub zasadzie Zaïtseva.
Czasami jednak termodynamicznie preferencyjnie powstaje alcena, mówimy o eliminacji Hofmanna. Dzieje się tak w szczególności, gdy zastosowana podstawa jest zatłoczona ( były. : Ktbuo) lub gdy masz jon amonu, który eliminuje na przykład. (Cząsteczka nosząca aminę w obecności MEI w liczbie równoważników wystarczających do utworzenia soli amonowej, a następnie wkładana w obecność mokrego i podgrzewanego tlenku srebra. Tworzenie alcene przez eliminację hofmanna i odlot z N (Me) 3 .)
Czynniki wpływające na reakcję [[[ modyfikator |. Modyfikator i kod ]
Parametry wpływające na eliminację E1 to:
- Wpływ R-: Kinetycznie decydującym krokiem jest szkolenie karbokacji (a nie jego reaktywność), więc im bardziej jest stabilny, tym bardziej faworyzuje się reakcję;
- Nucleoffuge: Połączenie linku Grupa C odejście Będąc kluczowym krokiem mechanizmu, reakcja silnie zależy od natury tego nukleoffingu. Im lepsza grupa początkowa, tym szybsza reakcja. Na przykład w przypadku odwodnienia alkoholi pierwszym krokiem jest protonowanie alkoholu do utworzenia oksonium, a zatem lepsza grupa początkowa (H (H. H 2 O ⇒ HO−). Un classement rapide des groupes partants serait :
R-SO 2 – (z r- ≡ me- lub ar-)> -iii – > -Br – > -Cl –
- (Symbol „>” oznacza: lepsze odejście grupy niż.)
Należy zauważyć, że podstawa nie wpływa na tego rodzaju reakcję rzędu 1, a to, o ile nie interweniuje podczas pierwszego kroku, który określa ogólną prędkość reakcji.
Grupa alkilowa halogené pochodnej [[[ modyfikator |. Modyfikator i kod ]
Mechanizm obejmuje pośrednik reakcji karbokacji. Jest to tym bardziej stabilne, ponieważ jest zastąpione. Zatem prędkość reakcji (zatem tworzenia karbokacji) przechodzi z pierwotnych pochodnych, wtórnych wówczas do trzeciego. W praktyce tylko te ostatnie i niektóre wtórne pochodne reagują z tym mechanizmem.
Nucleoft [[[ modyfikator |. Modyfikator i kod ]
Im bardziej polaryzowalne połączenie R-X, tym łatwiejsza rozpad. Ten link ma zatem zmniejszającą leczność („kruchość”) z fluoru do jodu: f> cl> I.
- (Symbol „>” oznacza: szybciej niż.)
Rozpuszczalnik [[[ modyfikator |. Modyfikator i kod ]
- Polaryzacja: rozpuszczalnik polarny stabilizuje karbokacja; Ułatwia to pierwszą reakcję. Szybkość wierzy z polaryjnością rozpuszczalnika.
- Protekcja: rozpuszczalnik dowodowy jest raczej upośledzonym: w rzeczywistości ryzykuje konkurując z karbokacją podczas reakcji kwasowo-zasadowej (silnej).
Zbieżność [[[ modyfikator |. Modyfikator i kod ]
Ta reakcja dotyczy nie tylko konkurencji z inną eliminacją, ale także z reakcjami podstawień nukleofilowych, w szczególności z S. N 1. Ten ostatni występuje w podobnych warunkach (oprócz ogrzewania).
Kinetyka E2 [[[ modyfikator |. Modyfikator i kod ]
Bymolekularna reakcja eliminacji ma globalną kinetykę 2 i rzędu częściowego 1 w odniesieniu do halogenowanego pochodnego R-X i podstawy B: – .
Reakcja odbywa się w jednym kroku, który jest bimolekularny.
Szybkość reakcji jest zgodna z prawem typu: W = k · [R-X] · [B: – ]
Lub :
- [R-X] reprezentuje stężenie halogenijącego pochodne (wyrażane w mol/l);
- [B: – ] reprezentuje podstawowe stężenie (wyrażone w mol/l);
- W reprezentuje prędkość reakcji eliminacji. Jest wyrażany w mol/s;
- k jest stałą prędkości. Jego jednostka, L 2 · Mol −1 ·S −1 , Dewuty od pozostałych trzech terminów.
Mechanizm E2 [[[ modyfikator |. Modyfikator i kod ]
Eliminacja bimolekularna jest reakcją na jeden krok, bimolekularny.

Podczas tego samego elementarnego etapu wystąpił owijanie protonu przez podstawę, tworzenie się podwójnego wiązania i odejście atomu halogenowego (nukleofuge) w postaci jonu falogennego. Mówimy o mechanizmie synchronicznym lub skoordynowanym.
Stereochemia [[[ modyfikator |. Modyfikator i kod ]
E2 jest reakcją stereospecyficzną, jeśli węgiel beta ma pojedynczy wodór. W odwrotnym przypadku może to być niesterospecyficzne. Wszystko zależy od podłoża.
Reakcja odbywa się tylko wtedy, gdy wodór i grupa początkowa znajdują się w pozycji antyperiplanarskiej.
Czynniki wpływające na reakcję [[[ modyfikator |. Modyfikator i kod ]
- Szybkość reakcji E2 zmienia się tylko słabo w zależności od klasy (pierwotna: I, wtórna: ii, trzeciorzęd: III) rozważanej flueniury. Niemniej jednak, im bardziej wysoka klasa, tym szybsza reakcja (III> II> i) z powodu sterycznego (eliminacja umożliwia odejście, w produkcie końcowym, obszerne podstawniki, a zatem „nieporęczne”, obecne w początkowym miejscu w początkowym Produkt, odczynnik, ponieważ wychodzimy z kąta Valenciela wynoszącego 109 ° 28 ‘, na poziomie hybrydyzowanego spinu 3 , niosąc proton H, pod kątem 120 ° Valenciel, w produkcie końcowym, w którym ten sam węgiel jest teraz hybrydyzowany odcinek 2 ). W tym przypadku na ogół faworyzujemy pierwotną halogenoalcan, ale kiedy jesteśmy w obecności wtórnego halogenoalcanu, różnicujemy reakcje typu E1 i E2 przez potencjalne efekty mezomeryczne („Mezomer” Efekt: ogólny termin określający odpowiadający efekt elektroniczny, aby uzyskać efekt elektroniczny, aby to zrobić Przeniesienie obciążenia, tutaj dodatnie, na grupie aryle, na przykład, bezpośredni sąsiad, a zatem „w α”, o połączeniu C-H, mówimy o „deprotonacji”, przez silną bazę). Każdy substytut łańcucha węglowego, który może ustabilizować jon węglowodanów (karbokacja C + ) Pośrednio według tego rodzaju efektu elektronicznego będzie promować mechanizm E1.
- Szybkość reakcji rośnie wraz z siłą podstawy, więc zajęłaby silną podstawę.
- Postać nukleofferu grupy początkowej ma również wpływ na reakcję, ale wpływ ten nie stanowi pierwotnego parametru, w przeciwieństwie do mechanizmu eliminacji jednorodkularnej (E1).
- Aby promować reakcyjny bimolekularny mechanizm eliminacji E2, mądrze jest zastosowanie rozpuszczalnika apotycznego (DMSO, THF, toluen, N -Hexan, itp. ).
Zbieżność [[[ modyfikator |. Modyfikator i kod ]
Ta reakcja dotyczy nie tylko konkurencji z E1, ale także z reakcjami podstawień nukleofilowych, w szczególności z S. N 2, które występują w podobnych warunkach.
Należy zauważyć, że reakcja eliminacji będzie wymagała więcej energii niż odpowiednia reakcja podstawienia, ogólnie poprzez ogrzewanie.
Zastosowana podstawa jest również nukleofil. To wyjaśnia konkurencję z reakcjami podstawowymi. Pod koniec reakcji można znaleźć produkty mieszane, eliminacyjne i substytucje.
Recent Comments