Catalyseur de Crabtree – Wikipedia wiki
Un article de Wikipédia, l’encyclopédie libre
 |
|
 |
|
| Des noms | |
|---|---|
| Nom IUPAC
( Sp -4) – (le 2 ,le 2 -Cycloocta-1,5-diène) (pyridine) (tricyclohexylphosphane) iridium (1+) hexafluoridophosphate (1−) |
|
| Identificateurs | |
| Chemspider | |
| Echa Infocard | 100.164.161 |
| QUELQUES | |
|
|
|
|
| Propriétés | |
| C trente et un H 50 F 6 IRNP 2 | |
| Masse molaire | 804.9026 g / mol |
| Apparence | Microcristaux jaunes |
| Point de fusion | 150 ° C (302 ° F; 423 K) (décomposer) [d’abord] |
|
Sauf lorsqu’il est indiqué autrement, des données sont données pour les matériaux à l’état standard (à 25 ° C [77 ° F], 100 kPa).
|
|
Composé chimique
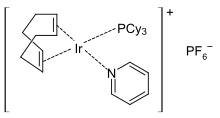
Catalyseur de Crabtree est un composé organariridium avec la formule [c 8 H douzième IRP (C 6 H 11 ) 3 C 5 H 5 N] pf 6 . Il s’agit d’un catalyseur homogène pour les réactions d’hydrogénation et de transfert d’hydrogène, développés par Robert H. Crabtree. Ce solide orange stable à l’air est disponible dans le commerce et connu pour son hydrogénation dirigée pour donner une stéréosélectivité trans avec le groupe de mise en scène. [2] [3]
Structure et synthèse [ modifier ]]
Le complexe a une géométrie moléculaire plane carrée, comme prévu pour A D 8 complexe. Il est préparé à partir du dimère de chlorure de cyclooctadiène iridium. [4]
Réactivité [ modifier ]]
Le catalyseur de Crabtree est efficace pour les hydrogénations des substrats mono-, di-, tri- et tétra-substitués. Alors que le catalyseur de Wilkinson et le catalyseur Schrock – Osborn ne catalysent pas l’hydrogénation d’une oléfine tétrasubstituée, le catalyseur de Crabtree le fait à des fréquences de renouvellement élevées (table). [2] [5]
-
Fréquences de roulement Substrat Catalyseur de Wilkinson Catalyseur Schrock – Osborn Catalyseur de Crabtree Hex-1-een 650 4000 6400 Cyclohexène 700 dix 4500 1-méthylcyclohexène 13 – 3800 2,3-diméthyl-mais-2-een – – 4000
Le catalyseur est réactif à température ambiante. [d’abord] La réaction est robuste sans séchage de solvants ni désoxygénation méticuleuse de l’hydrogène. Le catalyseur est tolérant des groupes fonctionnels faiblement basiques tels que l’ester, mais pas des alcools (voir ci-dessous) ou des amines. [2] Le catalyseur est sensible aux impuretés provoquant des protons. [6]
Le catalyseur devient irréversiblement désactivé après environ dix minutes à température ambiante, signalé par l’apparition d’une couleur jaune. Un processus de désactivation implique la formation de dimères à pont hydrure. [7] En conséquence, le catalyseur de Crabtree est généralement utilisé dans une charge de catalyseurs très faible.

Autres fonctions catalytiques: échange d’isotopes et isomérisation [ modifier ]]
Outre l’hydrogénation, le catalyseur catalyse l’isomérisation et l’hydroboration des alcènes. [d’abord]
Le catalyseur de Crabtree est utilisé dans les réactions d’échange d’isotopes. Plus précisément, il catalyse l’échange direct d’un atome d’hydrogène avec ses isotopes deutérium et son tritium, sans l’utilisation d’un intermédiaire. [8] Il a été démontré que l’échange d’isotopes avec le catalyseur de Crabtree est hautement régiosélectif. [9] [dix]
Influence de la réalisation de groupes fonctionnels [ modifier ]]
L’hydrogénation d’un terpen-4-ol démontre la capacité des composés avec des groupes de mise en scène (groupe –OH) à subir une hydrogénation diastéréosélective. Avec du palladium sur le carbone dans l’éthanol, la distribution du produit est de 20:80 en faveur du cis isomère ( 2B dans le schéma 1). Le côté polaire (avec le groupe hydroxyle) interagit avec le solvant. Cela est dû à une légère haptophilicité, un effet dans lequel un groupe fonctionnel se lie à la surface d’un catalyseur hétérogène et dirige la réaction. [11] [douzième] Dans le cyclohexane comme solvant, la distribution passe à 53:47 parce que l’haptophilicité n’est pas longue (il n’y a pas de groupe de mise en scène sur le cyclohexane). La distribution change complètement en faveur du trans isomère 2A Lorsque le catalyseur de Crabtree est utilisé dans le dichlorométhane. Cette sélectivité est à la fois prévisible et pratiquement utile. [13] Les groupes carbonyle sont également connus pour diriger l’hydrogénation par le catalyseur de Crabtree comme hautement régiosélectif. [14] [15] [16]
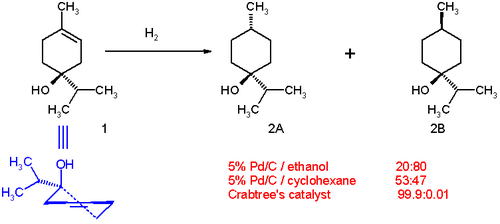
L’effet de mise en scène qui provoque la stéréosélectivité de l’hydrogénation de Terpen-4-OL avec le catalyseur de Crabtree est illustré ci-dessous.

Histoire [ modifier ]]
Crabtree et étudiant diplômé, George Morris, a découvert ce catalyseur dans les années 1970 tout en travaillant sur les analogues de l’Iridium du catalyseur basé sur le rhodium de Wilkinson à l’Institut de Chimie Des Substances Naturelles à Gif-sur-Yvette, près de Paris.
Les catalyseurs d’hydrogénation antérieurs comprenaient le catalyseur de Wilkinson et un complexe de rhodium cationique (I) avec deux groupes de phosphine développés par Osborn et Schrock. [17] Ces catalyseurs ont accompli l’hydrogénation par déplacement; Après addition d’hydrogène à travers le métal, un solvant ou un groupe de phosphine dissocié du rhodium métallique afin que l’oléfine soit hydrogénée puisse accéder au site actif. [2] Ce déplacement se produit rapidement pour les complexes de rhodium mais se produit à peine pour les complexes d’iridium. [18] Pour cette raison, la recherche à l’époque s’est concentrée sur les composés du rhodium au lieu des composés impliquant des métaux de transition de la troisième rangée, comme l’iridium. Wilkinson, Osborn et Schrock n’ont également utilisé que des solvants de coordination. [19]
Crabtree a noté que l’étape de dissociation du ligand ne se produit pas dans la catalyse hétérogène, et a ainsi posé que cette étape était limitante dans les systèmes homogènes. [2] Ils ont recherché des catalyseurs avec des «sites actifs créés de manière irréversible dans un solvant non coordonné». Cela a conduit au développement du catalyseur Crabtree et à l’utilisation du solvant ch 2 CL 2 .
Les références [ modifier ]]
- ^ un b c Crabtree, R. H. (2001). “(1,5-cyclooctadiène) (tricyclohexylphosphine) (pyridine) iridium (i) hexafluorophosphate”. E-EROS Encyclopedia of Reagents for Organic Synthesis . est ce que je: 10.1002 / 047084289x.rc290m.pub4 (inactif le 31 décembre 2022).
{{cite Encyclopedia}}: CS1 MAINT: DOI inactif en décembre 2022 (lien) - ^ un b c d C’est Robert H. Crabtree (1979). “Composés iridium dans la catalyse”. Acc. Chem. Res . douzième (9): 331–337. est ce que je: 10.1021 / ar50141a005 .
- ^ Brown, J. M. (1987). “Hydrogénation homogène dirigée”. Angew. Chem. Int. Éd. 26 (3): 190–203. est ce que je: 10.1002 / mai.198701901 .
- ^ Crabtree, R. H.; Morris, G. E. (1977). “Certains complexes de dioléfine de l’iridium (I) et un trans -Influence Series pour les complexes [ircl (COD) l] “. J. Organomet. Chem . 135 (3): 395–403. est ce que je: 10.1016 / s0022-328x (00) 88091-2 .
- ^ White, M. (2002-10-15). “Hydrogénation” (PDF) . Récupéré 2014-12-01 .
- ^ Xu, Yingjian; Mingos, D. Michael P.; Brown, John M. (2008). “Le catalyseur de Crabtree revisité; Effets du ligand sur la stabilité et la durabilité”. Chem. Comm. 2008 (2): 199–201. est ce que je: 10.1039 / b711979h . PMID 18092086 .
- ^ Crabtree, R.; Felkin, H.; Morris, G. (1977). “Les complexes cationiques de l’iridium dioléfine comme catalyseurs d’hydrogénation alcène et l’isolement de certains complexes hydrido apparentés”. Journal of Organometallic Chemistry . 141 (2): 205–215. est ce que je: 10.1016 / S0022-328X (00) 92273-3 .
- ^ Schou, S. (2009). “L’effet de l’ajout du catalyseur de Crabtree en noir de rhodium dans les réactions d’échange d’isotopes d’hydrogène direct”. Journal des composés étiquetés et radiopharmaceutiques . 52 (9): 376–381. est ce que je: 10.1002 / jlcr.1612 .
- ^ Valsborg, J.; Sorensen, L.; Foged, C. (2001). “L’échange d’isotopes d’hydrogène catalysés organiridium des dérivés de benzamide”. Journal des composés étiquetés et radiopharmaceutiques . 44 (3): 209–214. est ce que je: 10.1002 / jlcr.446 .
- ^ Hesk, D.; DAS, P.; Evans, B. (1995). “Dorisation des acétanilides et autres aromatiques substitués en utilisant [IR (COD) (CY 3 P) (py)] pf 6 comme catalyseur “. Journal des composés étiquetés et radiopharmaceutiques . 36 (5): 497–502. est ce que je: 10.1002 / jlcr.2580360514 .
- ^ Thompson, H.; Naipawer, R. (1973). “Contrôle stéréochimique des réductions. III. Approche des haptophilicités de groupe”. Journal de l’American Chemical Society . 95 (19): 6379–6386. est ce que je: 10.1021 / ja00800a036 .
- ^ Rowlands, G. (2002-01-01). “Hydrogénation” (PDF) . Récupéré 2014-12-01 .
- ^ Brown, J. (1987). “Hydrogénation homogène dirigée [nouvelles méthodes synthétiques (65)]”. Angew. Chem. Int. Éd. Engl. 26 (3): 190–203. est ce que je: 10.1002 / mai.198701901 .
- ^ Schultz, A.; McCloskey, P. (1985). “Le groupe carboxamide et carbalkoxy a dirigé des hydrogénations homogènes catalysées par l’iridium stéréosélectives”. Journal of Organic Chemistry . 50 (26): 5905–5907. est ce que je: 10.1021 / j00350a105 .
- ^ Crabtree, R. H.; Davis, M. W. (1986). “Diriger les effets dans l’hydrogénation homogène avec [IR (COD) (PCY3) (PY)] PF6”. J. Org. Chem . 51 (14): 2655–2661. est ce que je: 10.1021 / jo00364a007 .
- ^ Crabtree, R.; Davis, M. (1983). “Occurrence et origine d’un effet de mise en scène prononcé d’un groupe hydroxyle en hydrogénation avec [Ir (COD) P (C 6 H 11 ) 3 (py)] pf 6 “. Organométallique . 2 (5): 681–682. est ce que je: 10.1021 / OM00077A019 .
- ^ Schrock, R.; Osborn, J. A. (1976). “Hydrogénation catalytique en utilisant des complexes de rhodium cationiques. I. Évolution du système catalytique et hydrogénation des oléfines”. Journal de l’American Chemical Society . 98 (8): 2134-2143. est ce que je: 10.1021 / ja00424a020 .
- ^ Osborn, J.; Shapley, J. (1970). “Réarrangements intramoléculaires rapides dans les composés métalliques de transition pentacoordonnés. Mécanisme de réarrangement de certains complexes de fluxion iridium (I)”. Journal de l’American Chemical Society . 92 (23): 6976–6978. est ce que je: 10.1021 / ja00726a047 .
- ^ Young, J.; Wilkinson, G. (1966). “La préparation et les propriétés des Tris (triphénylphosphine) halogénorhodium (I) et certaines réactions, y compris l’hydrogénation homogène catalytique des oléfines et des acétylènes et leurs dérivés”. J. Chem. Soc. UN . 1966 : 1711. doi: 10.1039 / j196660001711 .
Recent Comments