Hémoglobín e – wikipedia wiki
Un article de Wikipédia, l’encyclopédie libre
Condition médicale
| Maladie de l’hémoglobine E | |
|---|---|
| Autres noms | Hémoglobine e |
 |
|
| Structure cristalline de l’hémoglobine E Mutant (Glu26Lys) Entrée PDB 1 . Chaîne alpha en chaîne rose, bêta en rouge. La mutation de la lysine a mis en évidence les sphères blanches. | |
| Spécialité | Hématologie |
Hémoglobine e ( Hbe ) est une hémoglobine anormale avec une mutation ponctuelle unique dans la chaîne β. À la position 26, il y a un changement dans l’acide aminé, de l’acide glutamique à la lysine (E26K). L’hémoglobine E est très courante chez les personnes d’origine sud-est de l’Asie, de l’Inde du Nord-Est, de Sri Lanka et du Bangladesh. [d’abord] [2]
La mutation βE affecte l’expression du gène β créant un site d’épissage alternatif dans l’ARNm aux codons 25-27 du gène β-globine. Grâce à ce mécanisme, il existe une légère carence en ARNm β normal et la production de petites quantités d’ARNm β anormal. La synthèse réduite de la chaîne β peut provoquer la β-thalassémie. De plus, cette variante d’hémoglobine a une union faible entre l’α- et la β-globine, provoquant l’instabilité lorsqu’il y a une grande quantité d’oxydant. [3] HBE peut être détecté sur électrophorèse.
Maladie de l’hémoglobine E (EE) [ modifier ]]

La maladie de l’hémoglobine E résulte lorsque la progéniture hérite du gène de HBE des deux parents. À la naissance, les bébés homozygotes pour l’hémoglobine E allèle ne présentent pas de symptômes car ils ont encore du HBF (hémoglobine fœtale). Au cours des premiers mois de la vie, l’hémoglobine fœtale disparaît et la quantité d’hémoglobine E augmente, de sorte que les sujets commencent à avoir une β-thalassémie légère.
Les sujets homozygotes pour l’hémoglobine E (deux allèles anormaux) ont une légère anémie hémolytique et une légère élargissement de la rate.
Il a essayé il a essayé: il était naunayuisy pour l’igname) [ modifier ]]
L’AE hétérozygote se produit lorsque le gène de l’hémoglobine E est hérité d’un parent et du gène de l’hémoglobine A de l’autre. C’est ce qu’on appelle le trait d’hémoglobine, et ce n’est pas une maladie. Les personnes qui ont un trait d’hémoglobine (hétérozygote) sont asymptomatiques et leur état n’entraîne généralement pas de problèmes de santé. Ils peuvent avoir un faible volume corpusculaire moyen (MCV) et des globules rouges très anormaux (cellules cibles), mais la pertinence clinique est principalement due au potentiel de transmission de l’E ou de la β-thalassémie. [4]
Maladie de la faucille-hémoglobine E (SE) [ modifier ]]
Les hétérozygotes composés atteints d’une maladie de la drépandia-hémoglobine E résultent lorsque le gène de l’hémoglobine E est hérité d’un parent et du gène de l’hémoglobine S de l’autre. À mesure que la quantité d’hémoglobine fœtale diminue et que l’hémoglobine augmente, une légère anémie hémolytique apparaît au début du développement. Les patients atteints de cette maladie éprouvent certains des symptômes de l’anémie falciforme, notamment une anémie légère modérée, un risque accru d’infection et des crises de faucille douloureuses. [5]
Hémoglobine E / β-thalassaémie [ modifier ]]

Les personnes qui ont l’hémoglobine E / β-thalassémie ont hérité d’un gène pour l’hémoglobine E d’un parent et un gène pour la β-thalassémie de l’autre parent. L’hémoglobine E / β-thalassémie est une maladie grave et n’a toujours pas de remède universelle. Il affecte plus d’un million de personnes dans le monde. [6] Les symptômes de l’hémoglobine E / β-thalassémie varient mais peuvent inclure le retard de croissance, l’élargissement de la rate (splénomégalie) et le foie (hépatomégalie), l’ictère, les anomalies osseuses et les problèmes cardiovasculaires. [7] Le traitement recommandé dépend de la nature et de la gravité des symptômes et peut impliquer une surveillance étroite des niveaux d’hémoglobine, des suppléments d’acide folique et des transfusions sanguines potentiellement régulières. [7]
Il existe une variété de phénotypes en fonction de l’interaction de HBE et α-thalassémie. La présence de l’α-thalassémie réduit la quantité de HBE généralement trouvée dans les hétérozygotes HBE. Dans d’autres cas, en combinaison avec certaines mutations de thalassémie, il fournit une résistance accrue au paludisme ( A. falciparum ). [4] Cette maladie a été décrite pour la première fois par Virginia Minnich en 1954, qui en a découvert une prévalence élevée en Thaïlande et l’a initialement qualifiée de “anémie méditerranéenne”. [7]
Épidémiologie [ modifier ]]
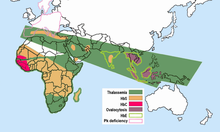
L’hémoglobine E est la plus répandue dans le continent en Asie du Sud-Est (Thaïlande, Myanmar, Cambodge, Laos, Vietnam [8] ), Sri Lanka, nord-est de l’Inde et du Bangladesh. En Asie du Sud-Est continentale, sa prévalence peut atteindre 30 ou 40%, et le nord-est de l’Inde, dans certaines zones, elle a des taux de transport qui atteignent 60% de la population. En Thaïlande, la mutation peut atteindre 50 ou 70%, et elle est plus élevée dans le nord-est du pays. Au Sri Lanka, il peut atteindre jusqu’à 40% et affecte ceux de la descendance cinghalaise et Vedda. [9] [dix] Il se trouve également aux hautes fréquences au Bangladesh et en Indonésie. [11] [douzième] Le trait peut également apparaître dans les personnes d’origine turque, chinoise et philippine. [d’abord] On estime que la mutation est apparue au cours des 5 000 dernières années. [13] En Europe, il y a eu des cas de familles atteintes d’hémoglobine E, mais dans ces cas, la mutation diffère de celle trouvée en Asie du Sud-Est. Cela signifie qu’il peut y avoir des origines différentes de la mutation βE. [14] [15]
Les références [ modifier ]]
- ^ un b “Hemoglobin E Trait – Encyclopédie Health – University of Rochester Medical Center” .
- ^ “Copie archivée” (PDF) . Archivé de l’original (PDF) le 2014-06-24 . Récupéré 2017-06-08 .
{{cite web}}: CS1 MAINT: Copie archivée comme titre (lien) - ^ Cheroff AI, Minnich V, Nanakorn S, et al. (1956). “Études sur l’hémoglobine E. I. Les caractéristiques cliniques, hématologiques et génétiques des syndromes de l’hémoglobine E”. J Lab Clin avec . 47 (3): 455–489. PMID 13353880 .
- ^ un b Bachir, D; Galacteros, F (novembre 2004), Maladie de l’hémoglobine E. (PDF) , Encyclopédie orpheline , récupéré 13 janvier, 2014
- ^ Département de la santé de l’Arkansas. “Fiche d’information sur la maladie de la faucille-hémoglobine” ” (PDF) .
- ^ Vichinsky E (2007). “Syndromes d’hémoglobine E” . Hematology Am Soc Hematol Educ Program . 2007 : 79–83. est ce que je: 10.1182 / ashedugation-2007.1.79 . PMID 18024613 . S2cid 10435042 .
- ^ un b c Fucharoen, Suthat; Weatherall, David J. (2012-08-01). “L’hémoglobine e thalassemias” . Perspectives de Cold Spring Harbour en médecine . 2 (8): A011734. deux: 10.1101 / cshPerSpect.A011734 . ISSN 2157-1422 . PMC 3405827 . PMID 22908199 .
- ^ Hémoglobine e trait , Université de Rochester Medical Center , récupéré 13 janvier, 2014
- ^ Sarkar, Jayanta; Ghosh, G. C. (2003). Populations des pays de la SAARC: perspectives bio-culturelles . ISBN 9788120725621 .
- ^ http://php.scripts.psu.edu/nxm2/1985%20Publications/1985-Roychoudhury-nei.pdf [ URL nue PDF ]]
- ^ Kumar, Dhavendra (2012-09-15). Troubles génétiques du sous-continent indien . ISBN 9781402022319 .
- ^ Olivieri NF, Pakbaz Z, Vichinsky et (2011). “Hb e / bêta-thalassaémie: un trouble commun et cliniquement divers” ” . Indian J. Med. Res . 134 (4): 522–31. PMC 3237252 . PMID 22089616 .
- ^ Ohashi; et al. (2004). “Disquilibre de liaison prolongée entourant la variante de l’hémoglobine E en raison de la sélection paludéenne” . Suis J Hum Genet . 74 (6): 1189–1208. est ce que je: 10.1086 / 421330 . PMC 1182083 . PMID 15114532 . Texte intégral gratuit
- ^ Kazazian HH, Jr., Waber PG, Boehm CD, Lee JI, Antonarakis SE, Fairbanks VF. (1984). “L’hémoglobine E chez les Européens: preuve supplémentaire de multiples origines du gène βE-Globine” . Suis J Hum Genet . 36 (1): 212–217. PMC 1684388 . PMID 6198908 .
{{cite journal}}: CS1 Maint: plusieurs noms: liste des auteurs (lien) Texte intégral gratuit - ^ Bain, Barbara J (juin 2006). Cellules sanguines: un guide pratique (4e éd.). Wiley-Blackwell. ISBN 978-1-4051-4265-6 .
Liens externes [ modifier ]]
Recent Comments