SNARE (Protein) – Wikipedia

SNARE-Proteine – “SNAP. REceptor “- sind eine große Proteinfamilie, die aus mindestens 24 Mitgliedern in Hefen und mehr als 60 Mitgliedern in Säugetierzellen besteht.[2][3] Die Hauptaufgabe von SNARE-Proteinen besteht darin, die Vesikelfusion zu vermitteln – die Fusion von Vesikeln mit der Zielmembran; Dies vermittelt insbesondere die Exozytose, kann aber auch die Fusion von Vesikeln mit membrangebundenen Kompartimenten (wie einem Lysosom) vermitteln. Die am besten untersuchten SNAREs sind diejenigen, die die Neurotransmitterfreisetzung von synaptischen Vesikeln in Neuronen vermitteln. Diese neuronalen SNAREs sind die Ziele der Neurotoxine, die für Botulismus und Tetanus verantwortlich sind, die von bestimmten Bakterien produziert werden.
SNAREs können in zwei Kategorien unterteilt werden: Vesikel oder v-SNAREs, die während des Knospens in die Membranen von Transportvesikeln eingebaut werden, und Ziel oder t-SNAREs, die mit Nervenendmembranen assoziiert sind. Es gibt Hinweise darauf, dass t-SNAREs stabile Subkomplexe bilden, die als Leitfaden für v-SNARE dienen und in die Membran eines proteinbeschichteten Vesikels eingebaut sind, um die Bildung des SNARE-Komplexes zu vervollständigen.[4] Mehrere SNARE-Proteine befinden sich sowohl auf Vesikeln als auch auf Zielmembranen. Daher berücksichtigt ein neueres Klassifizierungsschema strukturelle Merkmale von SNAREs und unterteilt sie in R-SNAREs und Q-SNAREs. Oft fungieren R-SNAREs als v-SNAREs und Q-SNAREs als t-SNAREs. R-SNAREs sind Proteine, die einen Arginin (R) -Rest zur Bildung der Nullionenschicht im zusammengesetzten Kern-SNARE-Komplex beitragen. Eine besondere R-SNARE ist Synaptobrevin, das sich in den synaptischen Vesikeln befindet. Q-SNAREs sind Proteine, die einen Glutamin (Q) -Rest zur Bildung der Nullionenschicht im zusammengesetzten Kern-SNARE-Komplex beitragen. Q-SNAREs umfassen Syntaxin und SNAP-25. Q-SNAREs werden abhängig von ihrer Position im Vier-Helix-Bündel weiter als Qa, Qb oder Qc klassifiziert.
Struktur[edit]
SNAREs sind kleine, häufig vorkommende, manchmal schwanzverankerte Proteine, die häufig posttranslational über eine C-terminale Transmembrandomäne in Membranen inseriert werden. Sieben der 38 bekannten SNAREs, einschließlich SNAP-25, haben keine Transmembrandomäne und sind stattdessen über Lipidmodifikationen wie Palmitoylierung an die Membran gebunden.[5] Schwanzverankerte Proteine können unter anderem in die Plasmamembran, das endoplasmatische Retikulum, die Mitochondrien und die Peroxisomen eingefügt werden, obwohl jede bestimmte SNARE auf eine einzigartige Membran abzielt. Das Targeting von SNAREs wird erreicht, indem entweder die Zusammensetzung der C-terminalen flankierenden Aminosäurereste oder die Länge der Transmembrandomäne verändert wird. Das Ersetzen der Transmembrandomäne durch Lipidanker führt zu einem Zwischenstadium der Membranfusion, in dem nur die beiden sich berührenden Blättchen verschmelzen und nicht die beiden distalen Blättchen der beiden Membrandoppelschichten.[6]
Obwohl sich SNAREs in Struktur und Größe erheblich unterscheiden, teilen sie alle ein Segment in ihrer cytosolischen Domäne, das als SNARE-Motiv bezeichnet wird und aus 60-70 Aminosäuren besteht und Heptad-Wiederholungen enthält, die die Fähigkeit haben, Coiled-Coil-Strukturen zu bilden. V- und t-SNAREs können reversibel zu engen Vier-Helix-Bündeln zusammengesetzt werden, die als “trans” -SNARE-Komplexe bezeichnet werden. In synaptischen Vesikeln bestehen die leicht gebildeten metastabilen “trans” -Komplexe aus drei SNAREs: Syntaxin 1 und SNAP-25, die in der Zellmembran vorhanden sind, und Synaptobrevin (auch als Vesikel-assoziiertes Membranprotein oder VAMP bezeichnet), das in der Vesikelmembran verankert ist.
Bei der neuronalen Exozytose sind Syntaxin und Synaptobrevin durch ihre C-terminalen Domänen in den jeweiligen Membranen verankert, während SNAP-25 über mehrere Cystein-verknüpfte Palmitoylketten an die Plasmamembran gebunden ist. Der Kern trans-SNARE-Komplex ist ein Vier-
-helix Bündel, wo man
 -helix Bündel, wo man
-helix Bündel, wo man -helix wird von Syntaxin 1, eins beigesteuert
-Helix von Synaptobrevin und zwei
-Helices werden von SNAP-25 beigesteuert.
Es wurde gezeigt, dass die in der Plasmamembran residenten SNAREs in verschiedenen Mikrodomänen oder Clustern vorhanden sind, deren Integrität für die exozytotische Kompetenz der Zelle wesentlich ist.
Membranfusion[edit]

Schichtung des Kern-SNARE-Komplexes. In der Mitte befindet sich die null hydrophile Ionenschicht, flankiert von hydrophoben Leucin-Zipper-Schichten.
Während der Membranfusion bilden v-SNARE- und t-SNARE-Proteine auf getrennten Membranen zusammen einen trans-SNARE-Komplex, der auch als “SNAREpin” bekannt ist. Abhängig vom Stadium der Fusion der Membranen können diese Komplexe unterschiedlich bezeichnet werden.
Während der Fusion von trans-SNARE-Komplexe, die Membranen verschmelzen und SNARE-Proteine, die an der Komplexbildung nach der Fusion beteiligt sind, werden dann als “cis“-SNARE-Komplex, weil sie jetzt in einem einzigen (oder cis) resultierende Membran. Nach der Fusion wird die cis-SNARE-Komplex wird durch ein Adapterprotein, Alpha-SNAP, gebunden und zerlegt. Dann katalysiert die als NSF bezeichnete hexamere ATPase (vom AAA-Typ) die ATP-abhängige Entfaltung der SNARE-Proteine und setzt sie zum Recycling in das Cytosol frei.
Es wird angenommen, dass SNAREs die wichtigsten erforderlichen Komponenten der Fusionsmaschinerie sind und unabhängig von zusätzlichen zytosolischen akzessorischen Proteinen funktionieren können. Dies wurde durch Engineering von “gespiegelten” SNAREs demonstriert, bei denen die SNARE-Domänen eher dem extrazellulären Raum als dem Cytosol zugewandt sind. Wenn Zellen, die v-SNAREs enthalten, Zellen kontaktieren, die t-SNAREs enthalten, trans-SNARE-Komplexe bilden sich und es kommt zur Zell-Zell-Fusion.[7]
Komponenten[edit]
Der Kern-SNARE-Komplex ist ein 4-
-helix-Bundle.[8] Synaptobrevin und Syntaxin tragen dazu bei
-helix jeweils, während SNAP-25 mit zwei teilnimmt
-helices (abgekürzt als Sn1 und Sn2). Die wechselwirkenden Aminosäurereste, die den SNARE-Komplex zippen, können in Schichten gruppiert werden. Jede Schicht hat 4 Aminosäurereste – jeweils einen Rest pro 4
-helices. Im Zentrum des Komplexes befindet sich das keine Ionenschicht Es besteht aus einem Arginin (R) – und drei Glutamin (Q) -Resten und wird von Leucin-Reißverschlüssen flankiert. Die Schichten ‘-1’, ‘+1’ und ‘+2’ in der Mitte des Komplexes folgen am ehesten der idealen Leucin-Reißverschluss-Geometrie und Aminosäure-Zusammensetzung.[9]
Das keine Ionenschicht besteht aus R56 aus VAMP-2, Q226 aus Syntaxin-1A, Q53 aus Sn1 und Q174 aus Sn2 und ist vollständig in den Leucin-Zipper-Schichten vergraben. Die positiv geladene Guanidinogruppe des Arginin (R) -Rests interagiert mit den Carboxylgruppen jedes der drei Glutamin (Q) -Reste.
Die flankierenden Leucin-Reißverschluss-Schichten wirken als wasserdichte Abdichtung, um die ionischen Wechselwirkungen vor dem umgebenden Lösungsmittel abzuschirmen. Belichtung der keine Ionenschicht Das Brechen des flankierenden Leucin-Reißverschlusses zum Wasserlösungsmittel führt zur Instabilität des SNARE-Komplexes und ist der mutmaßliche Mechanismus, durch den
-SNAP und NSF recyceln die SNARE-Komplexe nach Abschluss der Exozytose der synaptischen Vesikel.
Mechanismus der Membranfusion[edit]
Versammlung[edit]
SNARE-Proteine müssen sich zusammensetzen trans-SNARE-Komplexe, um die Kraft bereitzustellen, die für die Vesikelfusion erforderlich ist. Die vier α-Helix-Domänen (jeweils eine aus Synaptobrevin und Syntaxin und zwei aus SNAP-25) bilden zusammen ein Coiled-Coil-Motiv. Der geschwindigkeitsbestimmende Schritt im Assemblierungsprozess ist die Assoziation der Syntaxin-SNARE-Domäne, da sie normalerweise in einem “geschlossenen” Zustand gefunden wird, in dem sie nicht mit anderen SNARE-Proteinen interagieren kann.[10] Wenn Syntaxin offen ist, transDie Bildung des SNARE-Komplexes beginnt mit der Assoziation der vier SNARE-Domänen an ihren N-Termini. Die SNARE-Domänen bilden ein Coiled-Coil-Motiv in Richtung der C-Termini ihrer jeweiligen Domänen.
Es wird angenommen, dass das SM-Protein Munc18 eine Rolle beim Aufbau des SNARE-Komplexes spielt, obwohl der genaue Mechanismus, nach dem es wirkt, noch diskutiert wird. Es ist bekannt, dass der Verschluss von Munc18 Syntaxin in einer geschlossenen Konformation sperrt, indem er an seine α-helikalen SNARE-Domänen bindet, wodurch Syntaxin daran gehindert wird, in SNARE-Komplexe einzudringen (wodurch die Fusion gehemmt wird).[10] Der Verschluss ist jedoch auch in der Lage, das gesamte Vier-Helix-Bündel des zu binden trans-SNARE-Komplex. Eine Hypothese legt nahe, dass der Munc18-Verschluss während des Zusammenbaus des SNARE-Komplexes geschlossenes Syntaxin freisetzt, mit dem N-terminalen Peptid des Syntaxins assoziiert bleibt (was die Assoziation der Syntaxin-SNARE-Domäne mit anderen SNARE-Proteinen ermöglicht) und sich dann wieder an die neu gebildeten vier anschließt -helix SNARE-Komplex.[11] Dieser mögliche Mechanismus der Dissoziation und der anschließenden erneuten Assoziation mit den SNARE-Domänen könnte calciumabhängig sein.[12] Dies unterstützt die Idee, dass Munc18 eine wichtige regulatorische Rolle bei der Vesikelfusion spielt; Unter normalen Bedingungen wird die Bildung des SNARE-Komplexes durch Munc18 verhindert. Wenn der Munc18 jedoch ausgelöst wird, hilft er tatsächlich beim Aufbau des SNARE-Komplexes und wirkt dadurch als Fusionskatalysator.[11]
Reißverschluss und Fusionsporenöffnung[edit]

Die Membranfusion ist eine energetisch anspruchsvolle Reihe von Ereignissen, die die Translokation von Proteinen in der Membran und die Zerstörung der Lipiddoppelschicht sowie die Reformation einer stark gekrümmten Membranstruktur erfordert. Der Prozess des Zusammenbringens zweier Membranen erfordert Eingangsenergie, um die abstoßenden elektrostatischen Kräfte zwischen den Membranen zu überwinden. Der Mechanismus, der die Bewegung membranassoziierter Proteine von der Membrankontaktzone vor der Fusion weg reguliert, ist unbekannt, aber es wird angenommen, dass die lokale Zunahme der Membrankrümmung zu diesem Prozess beiträgt. SNAREs erzeugen Energie durch Protein-Lipid- und Protein-Protein-Wechselwirkungen, die als treibende Kraft für die Membranfusion wirken.
Ein Modell vermutet, dass die Kraft, die erforderlich ist, um zwei Membranen während der Fusion zusammenzubringen, von der Konformationsänderung in herrührt trans-SNARE-Komplexe bilden cis-SNARE-Komplexe. Die aktuelle Hypothese, die diesen Prozess beschreibt, wird als SNARE “Zippering” bezeichnet.[13]
Wenn der trans-SNARE-Komplex wird gebildet, die SNARE-Proteine befinden sich immer noch auf gegenüberliegenden Membranen. Wenn sich die SNARE-Domänen spontan weiter wickeln, bilden sie ein viel engeres und stabileres Vier-Helix-Bündel. Während dieses “Reißverschlusses” des SNARE-Komplexes wird angenommen, dass ein Teil der durch die Bindung freigesetzten Energie als molekulare Biegespannung in den einzelnen SNARE-Motiven gespeichert wird. Es wird postuliert, dass diese mechanische Spannung in den halbstarren Linkerregionen zwischen den Transmembrandomänen und dem SNARE-Helixbündel gespeichert wird.[14][15] Die energetisch ungünstige Biegung wird minimiert, wenn sich der Komplex peripher zum Ort der Membranfusion bewegt. Infolgedessen überwindet der Abbau der Spannung die Abstoßungskräfte zwischen dem Vesikel und der Zellmembran und drückt die beiden Membranen zusammen.[16]
Es wurden mehrere Modelle vorgeschlagen, um den nachfolgenden Schritt – die Bildung von Stiel und Fusionsporen – zu erklären. Die genaue Art dieser Prozesse bleibt jedoch umstritten. In Übereinstimmung mit der “Zipper” -Hypothese übt das sich verschärfende Helixbündel bei der Bildung des SNARE-Komplexes eine Torsionskraft auf die Domänen der Transmembrandomänen (TM) von Synaptobrevin und Syntaxin aus.[17] Dies bewirkt, dass die TM-Domänen innerhalb der getrennten Membranen kippen, wenn sich die Proteine enger wickeln. Die instabile Konfiguration der TM-Domänen führt schließlich dazu, dass die beiden Membranen fusionieren und die SNARE-Proteine innerhalb derselben Membran zusammenkommen, was als “cis“-SNARE-Komplex.[18] Infolge der Lipidumlagerung öffnet sich eine Fusionspore und lässt den chemischen Inhalt des Vesikels in die äußere Umgebung gelangen.
Die Kontinuumserklärung der Stielbildung legt nahe, dass die Membranfusion mit einem infinitesimalen Radius beginnt, bis sie sich radial zu einer stielartigen Struktur ausdehnt. Eine solche Beschreibung berücksichtigt jedoch nicht die Molekulardynamik von Membranlipiden. Neuere molekulare Simulationen zeigen, dass die Lipide durch die Nähe der Membranen gespreizt werden können, wobei eine Population von Lipiden ihre hydrophoben Schwänze in die benachbarte Membran einführt – wodurch effektiv ein “Fuß” in jeder Membran bleibt. Die Auflösung des gespreizten Lipidzustands erfolgt spontan unter Bildung der Stielstruktur. In dieser molekularen Ansicht ist der Zwischenzustand des gespreizten Lipids eher die geschwindigkeitsbestimmende Barriere als die Bildung des Stiels, der nun zum Minimum der freien Energie wird. Die energetische Barriere zur Herstellung der Konformation des gespreizten Lipids ist direkt proportional zum Abstand zwischen den Membranen. Die SNARE-Komplexe und ihr Zusammenpressen der beiden Membranen könnten daher die freie Energie liefern, die zur Überwindung der Barriere erforderlich ist.[19]
Demontage[edit]
Der Energieeinsatz, der für die SNARE-vermittelte Fusion erforderlich ist, stammt aus der Demontage des SNARE-Komplexes. Die vermutete Energiequelle ist der N-Ethylmaleimid-sensitive Faktor (NSF), eine ATPase, die an der Membranfusion beteiligt ist. NSF-Homohexamere binden und dissoziieren zusammen mit dem NSF-Cofaktor α-SNAP den SNARE-Komplex, indem sie den Prozess mit der ATP-Hydrolyse koppeln.[20] Dieser Prozess ermöglicht die Wiederaufnahme von Synaptobrevin zur weiteren Verwendung in Vesikeln, während die anderen SNARE-Proteine mit der Zellmembran assoziiert bleiben.
Die dissoziierten SNARE-Proteine haben einen höheren Energiezustand als die stabileren cis-SNARE-Komplex. Es wird angenommen, dass die Energie, die die Fusion antreibt, aus dem Übergang zu einer niedrigeren Energie stammt cis-SNARE-Komplex. Die ATP-Hydrolyse-gekoppelte Dissoziation von SNARE-Komplexen ist eine Energieinvestition, die mit dem “Spannen der Pistole” verglichen werden kann, so dass der Prozess nach Auslösung der Vesikelfusion spontan und mit optimaler Geschwindigkeit stattfindet. Ein vergleichbarer Prozess findet in Muskeln statt, in denen die Myosinköpfe zuerst ATP hydrolysieren müssen, um die notwendige Konformation für die Interaktion mit Aktin und den anschließenden Kraftschlag anzupassen.
Regulatorische Effekte auf die Exozytose[edit]
Regulation über SNAP-25-Palmitoylierung[edit]
Das Q-SNARE-Protein Synaptosomal-assoziiertes Protein 25 (SNAP-25) besteht aus zwei α-helikalen Domänen, die durch einen zufälligen Spulenlinker verbunden sind. Die zufällige Spulenlinkerregion ist am bemerkenswertesten für ihre vier Cysteinreste.[21] Die α-helikalen Domänen bilden zusammen mit denen von Syntaxin und Synaptobrevin (auch als Vesikel-assoziiertes Membranprotein oder VAMP bekannt) den 4-α-Helix-Coiled-Coil-SNARE-Komplex, der für eine effiziente Exozytose entscheidend ist.
Während Syntaxin und Synaptobrevin beide Transmembrandomänen enthalten, die das Andocken an Ziel- bzw. Vesikelmembranen ermöglichen, beruht SNAP-25 auf der Palmitoylierung von Cysteinresten, die in seiner zufälligen Spulenregion gefunden wurden, um an die Zielmembran anzudocken. Einige Studien haben gezeigt, dass die Assoziation mit Syntaxin über SNARE-Wechselwirkungen die Notwendigkeit solcher Docking-Mechanismen ausschließt. Syntaxin-Knockdown-Studien zeigten jedoch keine Abnahme des membrangebundenen SNAP-25, was darauf hindeutet, dass alternative Docking-Mittel existieren.[22] Die kovalente Bindung von Fettsäureketten an SNAP-25 über Thioesterbindungen mit einem oder mehreren Cysteinresten sorgt daher für eine Regulation des Andockens und letztendlich der SNARE-vermittelten Exozytose. Dieser Prozess wird durch ein spezialisiertes Enzym namens DHHC-Palmitoyltransferase vermittelt.[23] Es wurde auch gezeigt, dass die cysteinreiche Domäne von SNAP-25 schwach mit der Plasmamembran assoziiert ist, wodurch sie möglicherweise in der Nähe des Enzyms für die anschließende Palmitoylierung lokalisiert werden kann. Die Umkehrung dieses Prozesses wird von einem anderen Enzym namens Palmitoylprotein-Thioesterase durchgeführt (siehe Abbildung).

Es wird auch angenommen, dass die Verfügbarkeit von SNAP-25 im SNARE-Komplex möglicherweise räumlich durch Lokalisierung von Lipidmikrodomänen in der Zielmembran reguliert wird. Palmitoylierte Cysteinreste konnten über eine günstige Lipidumgebung (möglicherweise cholesterinreich), die zu den an die Cysteinreste von SNAP-25 gebundenen Fettsäureketten komplementär ist, in der gewünschten Zielmembranregion lokalisiert werden.[24]
SNAP-25-Regulation spannungsgesteuerter Ca2 + -Kanäle in neuronalen Axonterminals[edit]
Wenn ein Aktionspotential das Axonterminal erreicht, stimulieren Depolarisationsereignisse die Öffnung spannungsgesteuerter Calciumkanäle (VGCCs), die den schnellen Zufluss von Calcium entlang seines elektrochemischen Gradienten ermöglichen. Calcium stimuliert weiterhin die Exozytose durch Bindung an Synaptotagmin 1. Es wurde jedoch gezeigt, dass SNAP-25 die VGCC-Funktion in glutamatergen neuronalen Zellen negativ reguliert. SNAP-25 führt zu einer Verringerung der Stromdichte durch VGCCs und damit zu einer Verringerung der Calciummenge, die das Synaptotagmin bindet, was zu einer Verringerung der neuronalen glutamatergen Exozytose führt. Umgekehrt ermöglicht die Unterexpression von SNAP-25 eine Erhöhung der VGCC-Stromdichte und eine Erhöhung der Exozytose.[25]
Weitere Untersuchungen haben mögliche Zusammenhänge zwischen SNAP-25-Über- / Unterexpression und einer Vielzahl von Gehirnerkrankungen nahegelegt. Bei Aufmerksamkeitsdefizit- / Hyperaktivitätsstörung oder ADHS wurden Polymorphismen am SNAP-25-Genort beim Menschen mit der Krankheit in Verbindung gebracht, was auf eine mögliche Rolle bei ihrer Manifestation hinweist.[26] Dies wird weiter durch heterogene SNAP-25-Knockout-Studien nahegelegt, die an Colobom-mutierten Mäusen durchgeführt wurden und zu phänotypischen Eigenschaften von ADHS führten.[27] Studien haben auch eine Korrelation zwischen SNAP-25-Über- / Unterexpression und dem Auftreten von Schizophrenie gezeigt.[28][29]
Syntaxin und die Habc-Domäne[edit]
Syntaxin besteht aus einer Transmembrandomäne (TMD), einer alpha-helikalen SNARE-Domäne, einer kurzen Linkerregion und der Habc-Domäne, die aus drei alpha-helikalen Regionen besteht. Die SNARE-Domäne in Syntaxin dient als Zielstelle für das Andocken von SNAP-25 und Synaptobrevin, um das für den SNARE-Komplex und die anschließende Fusion erforderliche Vier-Helix-Bündel zu bilden. Die Habc-Domäne dient jedoch als autoinhibitorische Domäne in Syntaxin. Es wurde gezeigt, dass es sich umfaltet und mit der SNARE-Domäne von Syntaxin assoziiert, wodurch ein “geschlossener” Zustand induziert wird, wodurch eine physikalische Barriere für die Bildung des SNARE-Motivs entsteht. Umgekehrt kann sich die Habc-Domäne wieder von der SNARE-Domäne trennen, so dass Syntaxin frei ist, um sowohl mit SNAP-25 als auch mit Synaptobrevin zu assoziieren.[30]
Syntaxin 1B und leicht freisetzbarer Vesikelpool[edit]
Es gibt eine immense Vielfalt von Syntaxin-Subtypen mit 15 Sorten im menschlichen Genom.[31] Es wurde vermutet, dass Syntaxin1B eine Rolle bei der Regulierung der Anzahl von synaptischen Vesikeln spielt, die für die Exozytose im Axonterminal bereit sind. Dies wird auch als leicht freisetzbarer Pool (RRP) von Vesikeln bezeichnet. Eine Knock-out-Studie im Jahr 2014 zeigte, dass der Mangel an Syntaxin1B zu einer signifikanten Verringerung der RRP-Größe führte.[32]
Viele Neurotoxine wirken sich direkt auf SNARE-Komplexe aus. Toxine wie das Botulinum- und das Tetanus-Toxin wirken auf die SNARE-Komponenten. Diese Toxine verhindern ein ordnungsgemäßes Vesikelrecycling und führen zu einer schlechten Muskelkontrolle, Krämpfen, Lähmungen und sogar zum Tod.
Botulinum-Neurotoxin[edit]
Botulinumtoxin (BoNT) ist eines der wirksamsten Toxine, die jemals entdeckt wurden.[33] Es ist ein proteolytisches Enzym, das SNARE-Proteine in Neuronen spaltet. Seine Proteinstruktur besteht aus zwei Peptiduntereinheiten, einer schweren Kette (100 kDas) und einer leichten Kette (50 kDas), die durch eine Disulfidbindung zusammengehalten werden. Die Wirkung von BoNT folgt einem 4-Stufen-Mechanismus, der die Bindung an die neuronale Membran, Endozytose, Membrantranslokation und Proteolyse von SNARE-Proteinen umfasst.[34]
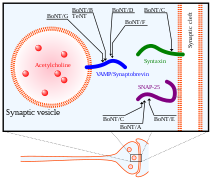
In ihrem Wirkungsmechanismus wird die schwere Kette von BoNT zunächst verwendet, um ihre neuronalen Ziele zu finden und an die Ganglioside und Membranproteine präsynaptischer Neuronen zu binden. Als nächstes wird das Toxin in die Zellmembran endozytiert. Die schwere Kette erfährt eine Konformationsänderung, die für die Translokation der leichten Kette in das Cytosol des Neurons wichtig ist. Nachdem die leichte Kette von BoNT in das Cytosol des Zielneurons gebracht wurde, wird sie schließlich aus der schweren Kette freigesetzt, so dass sie ihre aktiven Spaltstellen auf den SNARE-Proteinen erreichen kann.[34] Die leichte Kette wird durch Reduktion der Disulfidbindung, die die beiden zusammenhält, aus der schweren Kette gelöst. Die Reduktion dieser Disulfidbindung wird durch das NADPH-Thioredoxin-Reduktase-Thioredoxin-System vermittelt.[36] Die leichte Kette von BoNT wirkt als Metalloprotease auf SNARE-Proteinen, die von Zn (II) -Ionen abhängig ist.[37] spalten sie und beseitigen ihre Funktion bei der Exozytose.
Es sind 8 Isotypen von BoNT, BoNT / A – BoNT / H bekannt, die jeweils unterschiedliche spezifische Spaltstellen auf SNARE-Proteinen aufweisen. SNAP25, ein Mitglied der SNARE-Proteinfamilie, das sich in der Zellmembran befindet, wird durch die BoNT-Isotypen A, C und E gespalten. Die Spaltung von SNAP-25 durch diese BoNT-Isotypen hemmt ihre Funktion bei der Bildung des SNARE-Komplexes für die Fusion stark von Vesikeln zur synaptischen Membran. BoNT / C zielt auch auf Syntaxin-1 ab, ein weiteres SNARE-Protein, das sich in der synaptischen Membran befindet. Es degeneriert diese Syntaxin-Proteine mit einem ähnlichen Ergebnis wie bei SNAP-25. Ein drittes SNARE-Protein, Synaptobrevin (VAMP), befindet sich auf Zellvesikeln. VAMP2 wird von den BoNT-Isotypen B, D und F in synaptischen Neuronen angegriffen und gespalten.[33] Die Ziele dieser verschiedenen Isotypen von BoNT sowie Tetanus Neurotoxin (TeNT) sind in der Abbildung rechts dargestellt.
In jedem dieser Fälle verursacht Botulinum-Neurotoxin eine funktionelle Schädigung der SNARE-Proteine, was erhebliche physiologische und medizinische Auswirkungen hat. Durch die Schädigung von SNARE-Proteinen verhindert das Toxin, dass synaptische Vesikel mit der synaptischen Membran fusionieren und ihre Neurotransmitter in den synaptischen Spalt freisetzen. Mit der Hemmung der Neurotransmitterfreisetzung in den synaptischen Spalt können Aktionspotentiale nicht zur Stimulierung von Muskelzellen propagiert werden. Dies führt zu einer Lähmung der Infizierten und kann in schweren Fällen zum Tod führen. Obwohl die Wirkungen von Botulinum-Neurotoxin tödlich sein können, wurde es auch als therapeutisches Mittel bei medizinischen und kosmetischen Behandlungen verwendet.[38][39]
Tetanus-Neurotoxin[edit]

Tetanustoxin oder TeNT besteht aus einer schweren Kette (100 kDa) und einer leichten Kette (50 kDa), die durch eine Disulfidbindung verbunden sind. Die schwere Kette ist für die neurospezifische Bindung von TeNT an die Nervenendmembran, die Endozytose des Toxins und die Translokation der leichten Kette in das Cytosol verantwortlich. Die leichte Kette weist eine Zink-abhängige Endopeptidase- oder insbesondere Matrix-Metalloproteinase (MMP) -Aktivität auf, durch die die Spaltung von Synaptobrevin oder VAMP durchgeführt wird.[40]
Damit die leichte Kette von TeNT aktiviert werden kann, muss ein Zinkatom an jedes Toxinmolekül gebunden sein.[41] Wenn Zink gebunden ist, wird die Reduktion der Disulfidbindung hauptsächlich über das NADPH-Thioredoxinreduktase-Thioredoxin-Redoxsystem durchgeführt.[42] Dann ist die leichte Kette frei, um die Gln76-Phe77-Bindung von Synaptobrevin zu spalten.[40] Die Spaltung von Synaptobrevin beeinflusst die Stabilität des SNARE-Kerns, indem verhindert wird, dass dieser in die Konformation mit niedriger Energie eintritt, die das Ziel für die NSF-Bindung ist.[43] Diese Spaltung von Synaptobrevin ist das Endziel von TeNT, und selbst in niedrigen Dosen hemmt das Neurotoxin die Exozytose der Neurotransmitter.
Rolle bei der Freisetzung von Neurotransmittern[edit]
Neurotransmitter werden in leicht freisetzbaren Vesikelpools gespeichert, die im präsynaptischen Terminal eingeschlossen sind. Während der Neurosekretion / Exozytose spielen SNAREs eine entscheidende Rolle beim Andocken, Priming, der Fusion und der Synchronisation der Neurotransmitterfreisetzung in den synaptischen Spalt.
Der erste Schritt bei der synaptischen Vesikelfusion ist das Anbinden, bei dem die Vesikel aus dem Reservepool in physischen Kontakt mit der Membran gebracht werden. An der Membran ist Munc-18 zunächst in einer geschlossenen Struktur an Syntaxin 1A gebunden. Es wird postuliert, dass die Dissoziation von Munc-18 vom Komplex Syntaxin 1A freisetzt, um an die v-SNARE-Proteine zu binden.[44] Der nächste Schritt bei der Freisetzung ist das Andocken von Vesikeln, bei denen sich die v- und t-SNARE-Proteine vorübergehend auf kalziumunabhängige Weise verbinden. Die Vesikel werden dann vorbereitet, wobei die SNARE-Motive eine stabile Wechselwirkung zwischen dem Vesikel und der Membran bilden. Komplexine stabilisieren den vorbereiteten SNARE-Komplex und machen die Vesikel für eine schnelle Exozytose bereit.
Die Spanne der präsynaptischen Membran, die die vorbereiteten Vesikel und die dichte Sammlung von SNARE-Proteinen enthält, wird als aktive Zone bezeichnet. Spannungsgesteuerte Calciumkanäle sind stark um aktive Zonen konzentriert und öffnen sich als Reaktion auf die Membrandepolarisation an der Synapse. Der Zufluss von Kalzium wird durch Synaptotagmin 1 erfasst, das wiederum das Komplexinprotein verdrängt und es dem Vesikel ermöglicht, mit der präsynaptischen Membran zu fusionieren, um den Neurotransmitter freizusetzen. Es wurde auch gezeigt, dass die spannungsgesteuerten Calciumkanäle direkt mit den t-SNAREs-Syntaxin 1A und SNAP-25 sowie mit Synaptotagmin 1 interagieren. Die Wechselwirkungen können die Calciumkanalaktivität hemmen und die umliegenden Moleküle eng aggregieren die Release-Site.[45]
Es gab viele klinische Fälle, die SNARE-Gene mit neuralen Störungen verbinden. Bei einigen schizophrenen Patienten wurde im Hippocampusgewebe ein Mangel an SNAP-25-mRNA beobachtet, ein SNAP-25-Einzelnukleotid-Polymorphismus ist mit Hyperaktivität bei Autismus-Spektrum-Störungen verbunden, und eine Überexpression von SNAP-25B führt zum frühen Auftreten einer bipolaren Störung.[45]
Rolle bei der Autophagie[edit]
Makroautophagie ist ein katabolischer Prozess, bei dem doppelmembrangebundene Organellen, sogenannte Autophagosomen, gebildet werden, die den Abbau von Zellbestandteilen durch Fusion mit Lysosomen unterstützen. Während der Autophagie werden Teile des Zytoplasmas von einer becherförmigen Doppelmembranstruktur umgeben, die als Phagophor bezeichnet wird, und werden schließlich zum Inhalt des vollständig zusammengesetzten Autophagosoms. Die Biogenese von Autophagosomen erfordert die Initiierung und das Wachstum von Phagophoren, ein Prozess, von dem früher angenommen wurde, dass er durch die De-novo-Zugabe von Lipiden abläuft. Jüngste Erkenntnisse deuten jedoch darauf hin, dass die Lipide, die zu den wachsenden Phagophoren beitragen, aus zahlreichen Membranquellen stammen, darunter endoplasmatisches Retikulum, Golgi, Plasmamembran und Mitochondrien.[46] SNAREs spielen eine wichtige Rolle bei der Vermittlung der Vesikelfusion während der Initiierung und Expansion von Phagophoren sowie der Autophagosom-Lysosom-Fusion in den späteren Stadien der Autophagie.
Obwohl der Mechanismus der Phagophorinitiierung bei Säugetieren unbekannt ist, wurden SNAREs durch homotypische Fusion kleiner, mit Clathrin beschichteter Einzelmembranvesikel, die Atg16L, das v-SNARE VAMP7 und seinen Partner t-SNAREs: Syntaxin- enthalten, an der Phagophorbildung beteiligt 7, Syntaxin-8 und VTI1B.[47] In Hefen sind die t-SNAREs Sec9p und Sso2p für die Exozytose erforderlich und fördern das tubulovesikuläre Knospen von Atg9-positiven Vesikeln, die auch für die Biogenese von Autophagosomen erforderlich sind.[48][49] Das Ausschalten einer dieser SNAREs führt zur Akkumulation von kleinen Atg9-haltigen Vesikeln, die nicht fusionieren, wodurch die Bildung der präautophagosomalen Struktur verhindert wird.[49]
Neben der Phagophor-Assemblierung sind SNAREs auch wichtig für die Vermittlung der Autophagosom-Lysosom-Fusion. Bei Säugetieren sind die SNAREs VAMP7, VAMP8 und VTI1B für die Autophagosom-Lysosom-Fusion erforderlich. Dieser Prozess ist bei lysosomalen Speicherstörungen beeinträchtigt, bei denen sich Cholesterin im Lysosom ansammelt und SNAREs in cholesterinreichen Regionen der Membran bindet, wodurch deren Recycling verhindert wird.[50] Kürzlich wurde Syntaxin 17 (STX17) als autophagosomassoziiertes SNARE identifiziert, das mit VAMP8 und SNAP29 interagiert und für die Fusion mit dem Lysosom erforderlich ist.[51] STX17 ist auf der äußeren Membran von Autophagosomen lokalisiert, jedoch nicht auf Phagophoren oder anderen Autophagosomenvorläufern, wodurch verhindert wird, dass sie vorzeitig mit dem Lysosom fusionieren.[51] In Hefen erfordert die Fusion von Autophagosomen mit Vakuolen (dem Hefeäquivalent von Lysosomen) SNAREs und verwandte Proteine wie das Syntaxin-Homolog Vam3, das SNAP-25-Homolog Vam7, die Ras-ähnliche GTPase Ypt7 und das NSF-Ortholog Sec18.[46]
Verweise[edit]
- ^ Georgiev, Danko D; James F. Glazebrook (2007). “Subneuronale Informationsverarbeitung durch Einzelwellen und stochastische Prozesse”. In Lyshevski, Sergey Edward (Hrsg.). Handbuch zur Nano- und Molekularelektronik. Nano- und Microengineering-Serie. CRC Drücken Sie. S. 17–1–17–41. doi:10.1201 / 9781420008142.ch17 (inaktiv 2020-12-04). ISBN 978-0-8493-8528-5.CS1-Wartung: DOI ab Dezember 2020 inaktiv (Link)
- ^ Burri, Lena; Lithgow, Trevor (2004-01-01). “Ein vollständiger Satz von SNAREs in Hefe”. Der Verkehr. 5 (1): 45–52. doi:10.1046 / j.1600-0854.2003.00151.x. ISSN 1398-9219. PMID 14675424. S2CID 45480417.
- ^
Gerald K (2002). Zell- und Molekularbiologie (4. Aufl.). John Wiley & Sons. - ^ Malsam J, Söllner TH (1. Oktober 2011). “Organisation von SNAREs innerhalb des Golgi-Stacks”. Cold Spring Harbor Perspektiven in der Biologie. 3 (10): a005249. doi:10.1101 / cshperspect.a005249. PMC 3179334. PMID 21768609.
- ^ Hong W, Lev S (Januar 2014). “Anbindung der SNARE-Komplexe”. Trends in der Zellbiologie. 24 (1): 35–43. doi:10.1016 / j.tcb.2013.09.006. PMID 24119662.
- ^ Martens S, McMahon HT (21. Mai 2008). “Mechanismen der Membranfusion: unterschiedliche Akteure und gemeinsame Prinzipien”. Nature Reviews Molekulare Zellbiologie. 9 (7): 543–556. doi:10.1038 / nrm2417. PMID 18496517. S2CID 706741.
- ^ Hu C, Ahmed M, Melia TJ, Söllner TH, Mayer T, Rothman JE (13. Juni 2003). “Fusion von Zellen durch gespiegelte SNAREs”. Wissenschaft. 300 (5626): 1745–1749. doi:10.1126 / science.1084909. PMID 12805548. S2CID 18243885.
- ^
Sutton RB, Fasshauer D., Jahn R., Brunger AT (1998). “Kristallstruktur eines SNARE-Komplexes, der an der synaptischen Exozytose beteiligt ist, bei einer Auflösung von 2,4 Å”. Natur. 395 (6700): 347–353. doi:10.1038 / 26412. PMID 9759724. S2CID 1815214. - ^ Fasshauer D., Sutton RB, Brunger AT, Jahn R. (1998). “Konservierte Strukturmerkmale des synaptischen Fusionskomplexes: SNARE-Proteine, klassifiziert als Q- und R-SNAREs”. Verfahren der Nationalen Akademie der Wissenschaften. 95 (26): 15781–15786. doi:10.1073 / pnas.95.26.15781. PMC 28121. PMID 9861047.
- ^ ein b Burkhardt P., Hattendorf DA, Weis WI, Fasshauer D. (2008). “Munc18a steuert die SNARE-Assemblierung durch seine Wechselwirkung mit dem Syntaxin-N-Peptid”. EMBO J.. 27 (7): 923–33. doi:10.1038 / emboj.2008.37. PMC 2323264. PMID 18337752.
- ^ ein b Südhof TC, Rothman JE (Januar 2009). “Membranfusion: Auseinandersetzung mit SNARE- und SM-Proteinen”. Wissenschaft. 323 (5913): 474–7. doi:10.1126 / science.1161748. PMC 3736821. PMID 19164740.
- ^ Jahn R, Fasshauer D (2012). “Molekulare Maschinen zur Steuerung der Exozytose synaptischer Vesikel”. Natur. 490 (7419): 201–7. doi:10.1038 / nature11320. PMC 4461657. PMID 23060190.
- ^ Chen YA, Scheller RH (2001). “SNARE-vermittelte Membranfusion”. Nat. Rev. Mol. Cell Biol. 2 (2): 98–106. doi:10.1038 / 35052017. PMID 11252968. S2CID 205012830.
- ^ Wang Y., Dulubova I., Rizo J., Südhof TC (2001). Funktionsanalyse konservierter Strukturelemente in Hefesyntaxin Vam3p. J. Biol. Chem. 276 (30): 28598–605. doi:10.1074 / jbc.M101644200. PMID 11349128.
- ^ Kiessling V, Tamm LK (Januar 2003). “Messung von Abständen in unterstützten Doppelschichten mittels Fluoreszenzinterferenzkontrastmikroskopie: Polymerträger und SNARE-Proteine”. Biophysical Journal. 84 (1): 408–18. doi:10.1016 / s0006-3495 (03) 74861-9. PMC 1302622. PMID 12524294.
- ^ Risselada HJ, Kutzner C, Grubmüller H (2. Mai 2011). “Auf frischer Tat ertappt: Visualisierung von SNARE-vermittelten Fusionsereignissen im molekularen Detail”. ChemBioChem: Ein europäisches Journal für chemische Biologie. 12 (7): 1049–55. doi:10.1002 / cbic.201100020. hdl:11858 / 00-001M-0000-0027-C8EA-9. PMID 21433241. S2CID 14140718.
- ^ Fang Q, Lindau M (2014). “Wie könnten SNARE-Proteine eine Fusionspore öffnen?”. Physiologie. 29 (4): 278–85. doi:10.1152 / physiol.00026.2013. PMC 4103061. PMID 24985331.
- ^ Zucker, Robert S.; Kullmann, Dimitri M.; Kaeser, Pascal S. (August 2014). “Kapitel 15: Freisetzung von Neurotransmittern”. In Byrne, John H.; Heidelberger, Ruth; Waxham, M. Neal (Hrsg.). Von Molekülen zu Netzwerken: Eine Einführung in die zellulären und molekularen Neurowissenschaften. Akademische Presse. S. 443–488. ISBN 9780123982674.
- ^ Risselada HJ, Grubmüller H (April 2012). “Wie SNARE-Moleküle die Membranfusion vermitteln: aktuelle Erkenntnisse aus molekularen Simulationen”. Aktuelle Meinung in der Strukturbiologie. 22 (2): 187–96. doi:10.1016 / j.sbi.2012.01.007. hdl:11858 / 00-001M-0000-000F-9AF7-9. PMID 22365575.
- ^ Söllner T., Bennett MK, Whiteheart SW, Scheller RH, Rothman JE (1993). “Ein Protein-Assemblierungs-Disassemblierungs-Weg in vitro, der aufeinanderfolgenden Schritten des Andockens, der Aktivierung und der Fusion von synaptischen Vesikeln entsprechen kann”. Zelle. 75 (3): 409–18. doi:10.1016 / 0092-8674 (93) 90376-2. PMID 8221884. S2CID 26906457.
- ^ Bock, LV; Woodbury, DJ (9. August 2010). “Chemomechanische Regulation von SNARE-Proteinen, die mit molekulardynamischen Simulationen untersucht wurden”. Biophysical Journal. 99 (4): 1221–1230. doi:10.1016 / j.bpj.2010.06.019. PMC 2920728. PMID 20713006.
- ^ Greaves, Jennifer (5. April 2009). “Regulierung des SNAP-25-Handels und der SNAP-25-Funktion durch Palmitoylierung”. Transaktionen der Biochemical Society. 38 (Teil 1): 163–166. doi:10.1042 / BST0380163. PMID 20074052. S2CID 17636112.
- ^ Greaves, Jennifer (11. Mai 2010). Palmitoylierung der SNAP-25-Proteinfamilie: Spezifität und Regulation durch DHHC-Palmitoyltransferasen. Das Journal of Biological Chemistry. 285 (32): 24629–24638. doi:10.1074 / jbc.M110.119289. PMC 2915699. PMID 20519516.
- ^ Greaves, Jennifer (5. April 2009). “Regulierung des SNAP-25-Handels und der SNAP-25-Funktion durch Palmitoylierung”. Transaktionen der Biochemical Society. 38 (Teil 1): 163–166. doi:10.1042 / bst0380163. PMID 20074052. S2CID 17636112.
- ^ Condliffe, Steven B (3. Juni 2010). “Endogenes SNAP-25 reguliert native spannungsgesteuerte Calciumkanäle in glutamatergen Neuronen”. Das Journal of Biological Chemistry. 285 (32): 24968–24976. doi:10.1074 / jbc.M110.145813. PMC 2915732. PMID 20522554.
- ^ Corradini, Irene (21. Januar 2009). “SNAP-25 bei neuropsychiatrischen Störungen”. Annalen der New Yorker Akademie der Wissenschaften. 1152: 93–99. doi:10.1111 / j.1749-6632.2008.03995.x. PMC 2706123. PMID 19161380.
- ^ Hess, EJ (1992). Spontane lokomotorische Hyperaktivität in einer Mausmutante mit einer Deletion einschließlich des Snap-Gens auf Chromosom 2. Journal of Neuroscience. 12 (7): 2865–2874. doi:10.1523 / JNEUROSCI.12-07-02865.1992. PMC 6575838. PMID 1613559.
- ^ Thompson, PM (1998). “Veränderte Spiegel des synaptosomal assoziierten Proteins SNAP-25 bei Schizophrenie”. Biologische Psychiatrie. 43 (4): 239–243. doi:10.1016 / s0006-3223 (97) 00204-7. PMID 9513732. S2CID 20347660.
- ^ Gabriel, SM (1997). “Erhöhte Konzentrationen von präsynaptischen Proteinen im cingulären Cortex von Patienten mit Schizophrenie”. Archiv für Allgemeine Psychiatrie. 54 (6): 559–566. doi:10.1001 / archpsyc.1997.01830180077010. PMID 9193197.
- ^ MacDonald, Chris (3. April 2009). “Die Autoinhibition der komplexen SNARE-Assemblierung durch einen Konformationsschalter ist ein konserviertes Merkmal von Syntaxinen.”. Transaktionen der Biochemical Society. 38 (Pt 1): 209–212. doi:10.1042 / BST0380209. PMC 5242387. PMID 20074061.
- ^ Teng, Felicia Yu Hsuan (24. Oktober 2001). “Das Syntaxin”. Genombiologie. 2 (11): Übersichten 3012.1–7. doi:10.1186 / gb-2001-2-11-reviews3012. PMC 138984. PMID 11737951.
- ^ Mishima, Tatsuya (28. Februar 2014). “Syntaxin 1B, aber nicht Syntaxin 1A, ist für die Regulation der Exozytose synaptischer Vesikel und des leicht freisetzbaren Pools an zentralen Synapsen notwendig.”. PLUS EINS. 9 (2): e90004. doi:10.1371 / journal.pone.0090004. PMC 3938564. PMID 24587181.
- ^ ein b Peng L., Liu H., Ruan H., Tepp WH, Stoothoff WH, Brown RH, Johnson EA, Yao WD, Zhang SC, Dong M. (12. Februar 2013). “Die Zytotoxizität von Botulinum-Neurotoxinen zeigt eine direkte Rolle von Syntaxin 1 und SNAP-25 beim Überleben von Neuronen.”. Naturkommunikation. 4: 1472. doi:10.1038 / ncomms2462. PMC 4052923. PMID 23403573.
- ^ ein b Rossetto O., Pirazzini M., Bolognese P., Rigoni M., Montecucco C. (Dezember 2011). “Ein Update zum Wirkungsmechanismus von Tetanus- und Botulinum-Neurotoxinen” (PDF). Acta Chim Slov. 58 (4): 702–7. PMID 24061118.
- ^ Barr JR, Moura H., Boyer AE, Woolfitt AR, Kalb SR, Pavlopoulos A., McWilliams LG, Schmidt JG, Martinez RA, Ashley DL (2005). “Nachweis und Differenzierung von Botulinum-Neurotoxinen durch Massenspektrometrie”. Neu auftretende Infektion. Dis. 11 (10): 1578–83. doi:10.3201 / eid1110.041279. PMC 3366733. PMID 16318699.
- ^ Pirazzini M., Bordin F., Rossetto O., Shone CC, Binz T., Montecucco C. (Januar 2013). “Das Thioredoxinreduktase-Thioredoxin-System ist am Eintritt von Tetanus- und Botulinum-Neurotoxinen in das Cytosol von Nerventerminals beteiligt.” FEBS Briefe. 587 (2): 150–155. doi:10.1016 / j.febslet.2012.11.007. PMID 23178719. S2CID 207713815.
- ^ Silvaggi NR, Wilson D., Tzipori S., Allen KN (Mai 2008). “Katalytische Eigenschaften des Botulinum-Neurotoxins Eine leichte Kette, die durch die hochauflösende Struktur eines inhibitorischen Peptidkomplexes entdeckt wurde”. Biochemie. 47 (21): 5736–5745. doi:10.1021 / bi8001067. PMID 18457419.
- ^ Wheeler AH (1998). “Botulinumtoxin A, Zusatztherapie bei refraktären Kopfschmerzen im Zusammenhang mit perikranieller Muskelspannung”. Kopfschmerzen. 38 (6): 468–71. doi:10.1046 / j.1526-4610.1998.3806468.x. PMID 9664753. S2CID 12581048.
- ^ Garcia A, Fulton JE (1996). “Kosmetische Denervierung der Muskeln des Gesichtsausdrucks mit Botulinumtoxin. Eine Dosis-Wirkungs-Studie”. Dermatol Surg. 22 (1): 39–43. doi:10.1111 / j.1524-4725.1996.tb00569.x. PMID 8556256. S2CID 7703818.
- ^ ein b Schiavo G., Benfenati F., Poulain B., Rossetto O., Polverino de Laureto P., DasGupta BR, Montecucco C. (29. Oktober 1992). “Tetanus- und Botulinum-B-Neurotoxine blockieren die Neurotransmitterfreisetzung durch proteolytische Spaltung von Synaptobrevin”. Natur. 359 (6398): 832–5. doi:10.1038 / 359832a0. PMID 1331807. S2CID 4241066.
- ^ Schiavo G., Poulain B., Rossetto O., Benfenati F., Tauc L., Montecucco C. (Oktober 1992). “Tetanustoxin ist ein Zinkprotein und seine Hemmung der Neurotransmitterfreisetzung und der Proteaseaktivität hängt von Zink ab.”. Das EMBO Journal. 11 (10): 3577–83. doi:10.1002 / j.1460-2075.1992.tb05441.x. PMC 556816. PMID 1396558.
- ^ Pirazzini M., Bordin F., Rossetto O., Shone CC, Binz T., Montecucco C. (2013). “Das Thioredoxinreduktase-Thioredoxin-System ist am Eintritt von Tetanus- und Botulinum-Neurotoxinen in das Cytosol von Nerventerminals beteiligt.” FEBS Lett. 587 (2): 150–5. doi:10.1016 / j.febslet.2012.11.007. PMID 23178719. S2CID 207713815.
- ^ Pellegrini LL, O’Connor V., Lottspeich F., Betz H. (2. Oktober 1995). “Clostridiale Neurotoxine beeinträchtigen die Stabilität eines energiearmen SNARE-Komplexes, der die NSF-Aktivierung der synaptischen Vesikelfusion vermittelt.”. Das EMBO Journal. 14 (19): 4705–13. doi:10.1002 / j.1460-2075.1995.tb00152.x. PMC 394567. PMID 7588600.
- ^ Shi L., Kümmel D., Coleman J., Melia T. J., Giraudo CG (November 2011). “Die Doppelrolle von Munc18-1 beruht auf unterschiedlichen Bindungsmodi des zentralen Hohlraums mit dem Stx1A- und SNARE-Komplex.”. Molekularbiologie der Zelle. 22 (21): 4150–60. doi:10.1091 / mbc.e11-02-0150. PMC 3204075. PMID 21900493.
- ^ ein b Ramakrishnan NA, Drescher MJ, Drescher DG (Mai 2012). “Der SNARE-Komplex in neuronalen und sensorischen Zellen”. Molekulare und zelluläre Neurowissenschaften. 50 (1): 58–69. doi:10.1016 / j.mcn.2012.03.009. PMC 3570063. PMID 22498053.
- ^ ein b Moreau K., Ravikumar B., Renna M., Puri C., Rubinsztein DC (Juli 2011). “Die Reifung des Autophagosomenvorläufers erfordert eine homotypische Fusion”. Zelle. 146 (2): 303–317. doi:10.1016 / j.cell.2011.06.023. PMC 3171170. PMID 21784250.
- ^ Ravikumar B., Moreau K., Jahreiss L., Puri C., Rubinsztein DC (18. Juli 2010). “Die Plasmamembran trägt zur Bildung von präautophagosomalen Strukturen bei”. Naturzellbiologie. 12 (8): 747–757. doi:10.1038 / ncb2078. PMC 2923063. PMID 20639872.
- ^ Abeliovich, Hagai (1999). Das Zytoplasma zum Vakuolenhandel mit Aminopeptidase I erfordert einen t-SNARE / Sec1-Komplex, der aus Tlg2 und Vps45 besteht.. EMBO Journal. 18 (21): 6005–6016. doi:10.1093 / emboj / 18.21.6005. PMC 1171666. PMID 10545112.
- ^ ein b Nair U, Jotwani A, Geng J, Gammoh N, Richerson D, Yen WL, Griffith J, Nag S, Wang K, Moss T, Baba M, McNew JA, Jiang X, Reggiori F, Melia TJ, Klionsky DJ (Juli 2011) ). “SNARE-Proteine sind für die Makroautophagie erforderlich”. Zelle. 146 (2): 290–302. doi:10.1016 / j.cell.2011.06.022. PMC 3143362. PMID 21784249.
- ^ Fraldi A, Annunziata F, Lombardi A, Kaiser HJ, Medina DL, Spampanato C, Fedele AO, Polishchuk R, Sorrentino NC, Simons K, Ballabio A (24. September 2010). “Die lysosomale Fusion und die SNARE-Funktion werden durch die Cholesterinakkumulation bei lysosomalen Speicherstörungen beeinträchtigt.”. Das EMBO Journal. 29 (21): 3607–3620. doi:10.1038 / emboj.2010.237. PMC 2982760. PMID 20871593.
- ^ ein b Itakura E, Kishi-Itakura C, Mizushima N (Dezember 2012). “Das schwanzverankerte SNARE-Syntaxin 17 vom Haarnadeltyp zielt auf Autophagosomen zur Fusion mit Endosomen / Lysosomen ab.”. Zelle. 151 (6): 1256–1269. doi:10.1016 / j.cell.2012.11.001. PMID 23217709.
Externe Links[edit]
Recent Comments