Organogoldchemie – Wikipedia
Organogold-Chemie ist die Untersuchung von Verbindungen, die Gold-Kohlenstoff-Bindungen enthalten. Sie werden in der akademischen Forschung untersucht, haben aber sonst keine breite Anwendung gefunden. Die dominanten Oxidationsstufen für Organogoldverbindungen sind I mit der Koordinationszahl 2 und einer linearen Molekülgeometrie und III mit CN = 4 und einer quadratischen planaren Molekülgeometrie.[1][2][3] Die erste entdeckte Organogoldverbindung war Gold (I) carbid Au2C.2, die erstmals im Jahr 1900 vorbereitet wurde.[4]
Gold (I)[edit]
Gold (I) -Komplexe sind 2-koordinierte, lineare, diamagnetische 14-Elektronen-Spezies.[1][2][3] Sie existieren typischerweise als Addukte LAuR mit als Ligand L beispielsweise einem Triphenylphosphin oder einem Isocyanid. Der Ligand verhindert die Reduktion von Au (I) zu metallischem Au (0) unter Dimerisierung des organischen Rückstands. Gold (I) kann auch als das existieren aurate M.[AuR2] (der Ate-Komplex), wobei das Kation üblicherweise mit einem Komplexbildner ausgestattet ist, um die Stabilität zu verbessern. Der AuR2– – Anion ist ebenso linear wie andere M (d10) Arten wie Hg (Me)2 und Pd (ich)22+. Es ist bekannt, dass Gold Acetylide (die Polymerstrukturen bilden können), Carbene und Carbine bildet[citation needed]. Die klassische Methode zur Herstellung von LAuR-Verbindungen ist die Reaktion eines Grignard-Reagens mit einem Gold (I) -halogenid. Eine anschließende Reaktion mit einem Organolithium R-Li bildet den Ate-Komplex.
In einer speziellen Gruppe von Verbindungen fungiert ein Arylkohlenstoffatom als Brücke zwischen zwei Goldatomen. Eine solche Verbindung (MesAu)5entsteht bei einer Reaktion zwischen Au (CO) Cl und dem Mesityl Grignard. Kohlenstoff kann mit Gold bis zu einem Wert bis 6 koordiniert werden. Verbindungen vom Typ C (AuL)4 sind mit Methan und solchen vom Typ C (AuL) isolobal5+ isolobal mit dem Methanion. Diese hyperkoordinierten Organogoldcluster werden häufig durch aurophile Wechselwirkungen zwischen den formal geschlossenen Goldzentren stabilisiert.[5]
-
 Einige typische Organogoldarten mit verschiedenen Bindungsmodi.
Einige typische Organogoldarten mit verschiedenen Bindungsmodi.
Goldcyanidverbindungen (MAu (CN)2) sind für die Goldcyanidierung von Bedeutung, ein Verfahren zur Gewinnung von Gold aus minderwertigem Erz. Die Kohlenstoff-Metall-Bindung in Metallcyaniden ist normalerweise ionisch, es gibt jedoch Hinweise darauf, dass die C-Au-Bindung im Goldcyanidion kovalent ist.[6]
Gold (III)[edit]
Gold (III) -Komplexe sind 4-koordinierte, quadratische planare, diamagnetische, toxische 16-Elektronen-Spezies. Wenn die formale Koordinationszahl weniger als 4 beträgt, können Liganden wie Chlor dies durch Bildung eines Brückenliganden ausgleichen. Die intramolekulare Chelatbildung ist eine weitere Strategie. Im Allgemeinen sind Gold (III) -Verbindungen toxisch und daher weniger untersucht als Gold (I). Monoarylgold (III) -Komplexe sind eine gut untersuchte Klasse von Komplexen. Sie werden häufig durch direkte elektrophile Auration von Arenen durch AuCl hergestellt3.[7] Homoleptische Tetraalkylaurat (III) -Komplexe (z. B. Li[AuMe4]) sind auch gut charakterisiert.[8]
Goldkatalyse[edit]
Allgemeine Überlegungen[edit]
Goldkatalysierte Reaktionen fallen in zwei Hauptkategorien: heterogene Katalyse einschließlich Katalysatoren durch Goldnanopartikel (z. B. Au / TiO)2) und Thiol-Monoschicht-Goldoberflächen und Katalysatoren auf Aluminiumoxidträger, einschließlich Au / CeO auf Aluminiumoxidträger2. Diese Katalysatoren wurden für industriell wichtige Prozesse wie die Oxidation von Alkoholen, die Oxidation von Kohlenmonoxid (CO) und verschiedene selektive Hydrierungsreaktionen (z. B. Butadien zu Buten) untersucht. Obwohl sie oft effizient sind und nützliche oder einzigartige Selektivitäten aufweisen, besteht eine beträchtliche Unsicherheit hinsichtlich des Mechanismus von Prozessen, die durch verschiedene heterogene Goldkatalysatoren katalysiert werden, selbst im Vergleich zu anderen heterogenen Übergangsmetallkatalysatoren.
Im Gegensatz dazu werden bei der homogenen Katalyse mit Gold einfache oder ligandengebundene Gold (I) – oder Gold (III) -Verbindungen verwendet, die in organischen Lösungsmitteln löslich sind und zur Synthese von Feinchemikalien in der organischen Chemie verwendet werden.[9][10] Binäre Goldhalogenide und einfache Komplexe, einschließlich Gold (I) chlorid, Gold (III) chlorid und Chlorwasserstoffsäure, wurden als Komplexe eingesetzt. Diese Goldquellen führen jedoch schnell zu schlecht definierten und leicht zu deaktivierenden (durch Reduktion zu Au)0) aktive Katalysatoren in Lösung. Die Entwicklung gut definierter phosphin- oder NHC-ligierter Gold (I) -Komplexe war ein wichtiger Fortschritt und führte zu einem deutlichen Anstieg des Interesses an synthetischen Anwendungen der Goldkatalyse. Ligierte Gold (I) -Komplexe werden typischerweise als bankstabile (aber nicht reaktive) Chloride, LAuCl, hergestellt und gelagert, z. B. Chlor (triphenylphosphin) gold (I), die typischerweise durch Halogenidabstraktion mit Silbersalzen wie AgOTf, AgBF aktiviert werden4oder AgSbF6 eine kationische Gold (I) -Spezies zu erzeugen.[11][12] Obwohl der koordinativ ungesättigte Komplex “LAu+“wird fiktiv aus einem LAuCl / AgX-Gemisch erzeugt, die genaue Natur der kationischen Goldspezies und die Rolle des Silbersalzes bleiben etwas umstritten.[13][14][15] Das Abs-Nitrobenzoat, Bistriflimid und bestimmte Nitrilkomplexe stellen katalytisch aktive und dennoch isolierbare silberfreie Präkatalysatoren dar.
Kationisches Gold (I) bildet nach dem Dewar-Chatt-Duncanson-Modell π-Komplexe mit Alken- oder Alkinbindungen. Gold ist sicherlich nicht das einzige Metall, das diese Art von Bindung und Reaktivität aufweist, sondern auch mehrere Metallionen, die mit dem einfachen Proton isoliert sind (dh ein leeres s-Orbital): zum Beispiel Quecksilber (II) und Platin (II). Elektrophile Ionen und Komplexe wie diese mit einer starken Neigung zur Bildung von π-Komplexen sind allgemein bekannt als pi (π) -Säuren (siehe auch: Kation-Pi-Wechselwirkung).[16]
Gold (I) -Alken- und -Aalkinkomplexe sind elektrophil und anfällig für nukleophile Angriffe. Bei der Oxymercuration wird die resultierende Organomercurialspezies stöchiometrisch erzeugt und erfordert einen zusätzlichen Schritt zur Freisetzung des Produkts. Im Fall von Gold schließt die Protonolyse der Au-C-Bindung den Katalysezyklus und ermöglicht die Koordination eines anderen Substrats. Einige praktische Vorteile der Gold (I) -Katalyse sind: 1) Luftstabilität (aufgrund des hohen Oxidationspotentials von Au (I)), 2) Toleranz gegenüber zufälliger Feuchtigkeit (aufgrund seiner geringen Oxophilie) und 3) relativ geringe Toxizität im Vergleich zu andere Pi-Säuren (z. B. Pt (II) und Hg (II)). Chemisch gesehen werden Au (I) -Komplexe typischerweise nicht zu höheren Oxidationsstufen oxidiert, und Au (I) -Alkyle und -Vinyle sind nicht anfällig für die Eliminierung von β-Hydriden.[17]
-
 Typischer Mechanismus für die Gold (I) -katalysierte Hydrofunktionalisierung von Alkinen und Allenen.
Typischer Mechanismus für die Gold (I) -katalysierte Hydrofunktionalisierung von Alkinen und Allenen.
Historische Entwicklung[edit]
1976 berichteten Thomas und Mitarbeiter über die Umwandlung von Phenylacetylen in Acetophenon unter Verwendung von Tetrachloroaurinsäure in einer Ausbeute von 37%.[18] Bei dieser Reaktion wurde Gold (III) als homogener Katalysator verwendet, der Quecksilber bei der Oxyquecksilberung ersetzte. Dieselbe Studie listet eine veröffentlichte Ausbeute> 150% auf, was auf eine Katalyse hinweist, die von den Chemikern möglicherweise nicht anerkannt wurde.
1991 reagierte Utimoto mit Gold (III) (NaAuCl4) mit Alkinen und Wasser.[19] Teles identifizierte einen Hauptnachteil dieser Methode, da Au (III) schnell zu katalytisch totem metallischem Gold reduziert wurde und 1998 für dieselbe Umwandlung zum Thema des ligandengestützten Au (I) zurückkehrte:[20]
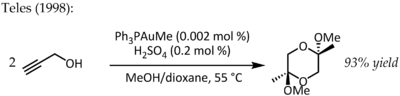
Diese besondere Reaktion zeigte eine fantastische katalytische Effizienz und würde in den kommenden Jahren eine Reihe von Forschungen zur Verwendung von Phosphingold (I) -Komplexen für die Aktivierung von CC-Mehrfachbindungen auslösen.[21] Trotz der geringeren Stabilität von Gold (III) -Komplexen unter katalytischen Bedingungen kann einfaches AuCl verwendet werden3 wurde in einigen Fällen auch als wirksamer Katalysator befunden. Zum Beispiel meldete Hashmi ein AuCl3-katalysierte Alkin / Furan-Diels-Alder-Reaktion – eine Art von Cycloaddition, die normalerweise nicht auftritt – zur Synthese von 2,3-disubstituierten Phenolen:[22]

Weitere mechanistische Studien kommen zu dem Schluss, dass dies keine konzertierte Transformation ist, sondern eine anfängliche Alkinhydroarylierung, gefolgt von einer Reihe nicht offensichtlicher intramolekularer Umlagerungen, die mit einer 6π-Elektrocyclisierung und -Rearomatisierung abgeschlossen werden.
Relativistische Effekte sind in der Organogoldchemie aufgrund der großen Kernladung des Metalls signifikant (Z. = 79). Infolge der relativistisch erweiterten 5d Orbitale kann das LAu-Fragment eine benachbarte Carbokation durch Elektronendonation ins Leere stabilisieren pOrbital vom Typ. Zusätzlich zu ihrer erwarteten carbokationsähnlichen Reaktivität weisen diese Kationen auch einen signifikanten Carbencharakter auf, eine Eigenschaft, die bei katalytischen Transformationen wie Cyclopropanierung und CH-Insertion ausgenutzt wurde.[23] Propargylester können als Vorläufer für kationische Gold-Vinylcarben-Zwischenprodukte dienen, die konzertiert mit Alkenen reagieren können, um das Cyclopropanierungsprodukt zu ergeben. Die Verwendung eines chiralen Liganden ((R.) -DTBM-SEGPHOS) führte zu guten bis ausgezeichneten Enantioselektivitäten.[24]

Obwohl Echavarren erstmals über die Herstellung von chiralen Bisphosphindigold (I) -Komplexen für die enantioselektive Goldkatalyse über den typischen pi-Aktivierungsmechanismus berichtete,[25] Ein frühes, atypisches Beispiel für die enantioselektive Katalyse durch Gold wurde 1986 von Hayashi und Ito beschrieben.[26] Bei diesem Verfahren werden Benzaldehyd und Methylisocyanoacetat in Gegenwart eines chiralen Ferrocenylphosphinliganden und eines Bis (isocyanid) gold (I) -Komplexes unter Bildung eines chiralen Oxazolins cyclisiert. Da Oxazoline unter Bildung eines 1,2-Aminoalkohols hydrolysiert werden können, ist diese Reaktion das erste Beispiel für eine katalytische, asymmetrische Aldolreaktion.
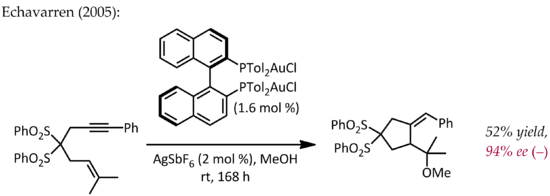
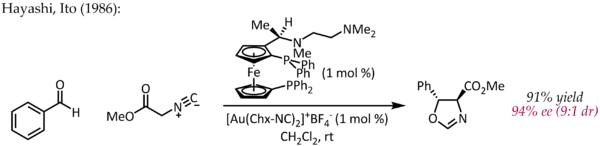
Im Gegensatz zu den anderen oben beschriebenen Reaktionen beinhaltet diese Reaktion keine Aktivierung einer CC-Doppel- oder Dreifachbindung durch Gold. In einem einfachen mechanistischen Bild koordiniert Gold (I) gleichzeitig an zwei Phosphinliganden und die Kohlenstoffisocyanatgruppe [27] welches dann von der Carbonylgruppe angegriffen wird. Weitere Studien zum Bindungsmodus von Au (I) zeigen, dass dieses einfache Bild möglicherweise überarbeitet werden muss.
Die heterogene Goldkatalyse ist eine ältere Wissenschaft. Gold ist aufgrund seiner Oxidationsstabilität und seiner unterschiedlichen Morphologie, beispielsweise Goldclustermaterialien, ein attraktives Metall. Es wurde gezeigt, dass Gold bei der CO-Oxidation bei niedriger Temperatur und der Acetylenhydrochlorierung zu Vinylchloriden wirksam ist. Die genaue Art der katalytischen Stelle bei dieser Art von Verfahren wird diskutiert.[28] Die Vorstellung, dass Gold eine Reaktion katalysieren kann, bedeutet nicht, dass dies der einzige Weg ist. Andere Metalle können jedoch die gleiche Arbeit kostengünstig erledigen, insbesondere in den letzten Jahren Eisen (siehe Organoiron-Chemie).
Goldkatalysierte Reaktionen[edit]
Obwohl Gold keine kommerzielle Bedeutung hat, katalysiert es viele organische Umwandlungen, üblicherweise die Bildung von Kohlenstoff-Kohlenstoff-Bindungen aus Au (I) und die Bildung von CX (X = O, N) -Bindungen aus dem Au (III) -Zustand aufgrund der härteren Lewis-Acidität dieses Ions . Während des letzten Jahrzehnts haben mehrere Studien gezeigt, dass Gold CC- und C-Heteroatom-Kreuzkupplungsreaktionen, die einen Au (I) / Au (III) -Zyklus durchlaufen, effizient katalysieren kann.[29] Hong C. Shen fasste homogene Reaktionen unter Bildung cyclischer Verbindungen in 4 Hauptkategorien zusammen:[30]

- Enincyclisierung, insbesondere Cycloisomerisierung, ein frühes Beispiel ist eine 5-exo-dig 1,6-Enincycloisomerisierung:[32]
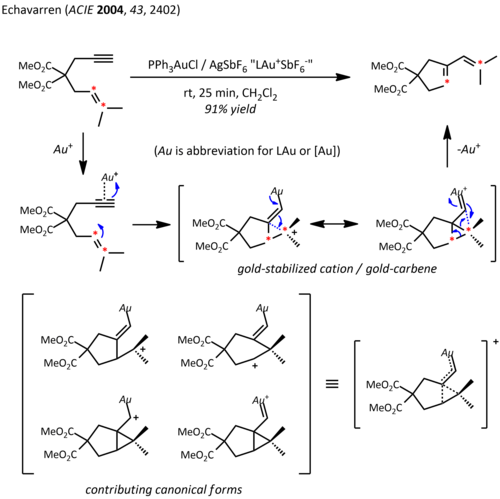
Andere Reaktionen sind die Verwendung von Gold bei der Aktivierung von CH-Bindungen[34] und Aldolreaktionen. Gold katalysiert auch Kupplungsreaktionen.[35]
Einschränkungen[edit]
Während goldkatalysierte Hydrofunktionalisierung von Alkinen, Allenen und Allylalkoholen[36] tritt leicht unter vergleichsweise milden Bedingungen auf, unaktiviertes Alken bleibt in den meisten Fällen schlechte Substrate,[37] zum großen Teil aufgrund der Beständigkeit der intermediären Alkylgold (I) -Komplexe gegen Protodeauration.[38] Die Entwicklung intermolekularer goldkatalysierter Transformationen blieb ebenfalls hinter der Entwicklung intramolekularer zurück.[39]
Verweise[edit]
- ^ ein b Elschenbroich, C. und Salzer, A. (1992) Organometallics: Eine kurze Einführung. Wiley-VCH: Weinheim. ISBN 3-527-28165-7
- ^ ein b Parish, RV (1997). “Organogoldchemie: II-Reaktionen”. Gold Bulletin. 30 (2): 55–62. doi:10.1007 / BF03214757.
- ^ ein b Parish, RV (1998). “Organogoldchemie: III-Anwendungen”. Gold Bulletin. 31: 14–21. doi:10.1007 / BF03215470.
- ^ Mathews, JA; Watters, LL (2002-05-01). “Das Hartmetall aus Gold”. Zeitschrift der American Chemical Society. 22 (2): 108–111. doi:10.1021 / ja02040a010.
- ^ Schmidbaur, Hubert; Schier, Annette (05.12.2011). “Aurophile Wechselwirkungen als Gegenstand aktueller Forschung: ein Update”. Bewertungen der Chemical Society. 41 (1): 370–412. doi:10.1039 / C1CS15182G. ISSN 1460-4744. PMID 21863191.
- ^ Wang, XB; Wang, YL; Yang, J.; Xing, XP; Li, J.; Wang, LS (2009). “Hinweise auf eine signifikante kovalente Bindung in Au (CN)2– –“. Zeitschrift der American Chemical Society. 131 (45): 16368–70. doi:10.1021 / ja908106e. PMID 19860420.
- ^ Kharasch, MS; Isbell, Horace S. (1931-08-01). “Die Chemie organischer Goldverbindungen. III. Direkte Einführung von Gold in den aromatischen Kern (vorläufige Mitteilung)”. Zeitschrift der American Chemical Society. 53 (8): 3053–3059. doi:10.1021 / ja01359a030. ISSN 0002-7863.
- ^ Rice, Gary W.; Tobias, R. Stuart. (1975-10-01). “Synthese von Tetramethylaurat (III). Strukturen von Lithiumdimethylaurat und Lithiumtetramethylaurat in Lösung”. Anorganische Chemie. 14 (10): 2402–2407. doi:10.1021 / ic50152a020. ISSN 0020-1669.
- ^ Goldkatalyse für die organische Synthese F. Dean Toste (Herausgeber) Thematische Reihe im Open Access Beilstein Journal of Organic Chemistry
- ^ Raubenheimer, HG; Schmidbaur, H. (2014). “Der späte Start und der erstaunliche Aufschwung in der Goldchemie”. Journal of Chemical Education. 91 (12): 2024–2036. Bibcode:2014JChEd..91.2024R. doi:10.1021 / ed400782p.
- ^ Ranieri, Beatrice; Escofet, Imma; Echavarren, Antonio M. (24.06.2015). “Anatomie der Goldkatalysatoren: Fakten und Mythen”. Org. Biomol. Chem. 13 (26): 7103–7118. doi:10.1039 / c5ob00736d. ISSN 1477-0539. PMC 4479959. PMID 26055272.
- ^ Wang, Yi-Ming; Lackner, Aaron D.; Toste, F. Dean (2013-11-14). “Entwicklung von Katalysatoren und Liganden für die enantioselektive Goldkatalyse”. Berichte über chemische Forschung. 47 (3): 889–901. doi:10.1021 / ar400188g. PMC 3960333. PMID 24228794.
- ^ Zhdanko, Alexander; Maier, Martin E. (09.09.2015). “Erklärung der” Silber-Effekte “bei der Gold (I) -katalysierten Hydroalkoxylierung von Alkinen”. ACS-Katalyse. 5 (10): 5994–6004. doi:10.1021 / acscatal.5b01493.
- ^ Homs, Anna; Escofet, Imma; Echavarren, Antonio M. (2013). “Über den Silbereffekt und die Bildung von Chlorid-verbrückten Digold-Komplexen”. Bio-Briefe. 15 (22): 5782–5785. doi:10.1021 / ol402825v. PMC 3833279. PMID 24195441.
- ^ Wang, Dawei; Cai, Rong; Sharma, Sripadh; Jirak, James; Thummanapelli, Sravan K.; Akhmedov, Novruz G.; Zhang, Hui; Liu, Xingbo; Petersen, Jeffrey L. (2012-05-18). “”“”Silber-Effekt “in der Gold (I) -Katalyse: Ein übersehener wichtiger Faktor”. Zeitschrift der American Chemical Society. 134 (21): 9012–9019. doi:10.1021 / ja303862z. PMID 22563621.
- ^ Fürstner, A.; Davies, PW (2007). “Katalytische carbophile Aktivierung: Katalyse durch Platin- und Gold-π-Säuren”. Angewandte Chemie International Edition. 46 (19): 3410–3449. doi:10.1002 / anie.200604335. PMID 17427893.
- ^ Shen, HC (2008). “Jüngste Fortschritte bei der Synthese von Heterocyclen und Carbocyclen durch homogene Goldkatalyse. Teil 1: Heteroatomaddition und Hydroarylierungsreaktionen von Alkinen, Allenen und Alkenen”. Tetraeder. 64 (18): 3885–3903. doi:10.1016 / j.tet.2008.01.081.
- ^ Norman, ROC; Parr, WJE; Thomas, CB (1976). “Die Reaktionen von Alkinen, Cyclopropanen und Benzolderivaten mit Gold (III)”. Zeitschrift der Chemical Society, Perkin Transactions 1 (18): 1983. doi:10.1039 / P19760001983.
- ^ Fukuda, Y.; Utimoto, K. (1991). “Effektive Umwandlung von nicht aktivierten Alkinen in Ketone oder Acetale mit einem Gold (III) -Katalysator”. Das Journal of Organic Chemistry. 56 (11): 3729–3731. doi:10.1021 / jo00011a058.
- ^ Teles, JH; Brode, S.; Chabanas, M. (1998). “Kationische Gold (I) -Komplexe: Hocheffiziente Katalysatoren für die Addition von Alkoholen an Alkine”. Angewandte Chemie International Edition. 37 (10): 1415–1418. doi:10.1002 / (SICI) 1521-3773 (19980605) 37:10<1415::AID-ANIE1415>3.0.CO; 2-N. PMID 29710887.
- ^ Nugent, WA (2012). “”“”Black Swan Events “in der organischen Synthese”. Angewandte Chemie International Edition. 51 (36): 8936–49. doi:10.1002 / anie.201202348. PMID 22893229.
- ^ Hashmi, ASK; Frost, TM; Bats, JW (2000). “Hochselektive goldkatalysierte Arensynthese”. Zeitschrift der American Chemical Society. 122 (46): 11553–11554. doi:10.1021 / ja005570d.
- ^ Gorin, David J.; Toste, F. Dean (2007). “Relativistische Effekte in der homogenen Goldkatalyse”. Natur. 446 (7134): 395–403. Bibcode:2007Natur.446..395G. doi:10.1038 / nature05592. PMID 17377576.
- ^ Johansson, Magnus J.; Gorin, David J.; Staben, Steven T.; Toste, F. Dean (30.11.2005). “Gold (I) -katalysierte stereoselektive Olefincyclopropanierung”. Zeitschrift der American Chemical Society. 127 (51): 18002–18003. doi:10.1021 / ja0552500. PMID 16366541.
- ^ Muñoz, M. Paz; Adrio, Javier; Carretero, Juan Carlos; Echavarren, Antonio M. (2005-02-12). “Ligandeneffekte bei der Gold- und Platinkatalysierten Cyclisierung von Eninen: Chirale Goldkomplexe für die enantioselektive Alkoxycyclisierung”. Metallorganika. 24 (6): 1293–1300. doi:10.1021 / om0491645.
- ^ Ito, Y.; Sawamura, M.; Hayashi, T. (1986). “Katalytische asymmetrische Aldolreaktion: Reaktion von Aldehyden mit Isocyanoacetat, katalysiert durch einen chiralen Ferrocenylphosphin-Gold (I) -Komplex”. Zeitschrift der American Chemical Society. 108 (20): 6405–6406. doi:10.1021 / ja00280a056.
- ^ Togni, A.; Pastor, SD (1990). “Chirale Kooperativität: Die Art des diastereoselektiven und enantioselektiven Schritts in der Gold (I) -katalysierten Aldolreaktion unter Verwendung chiraler Ferrocenylaminliganden”. Das Journal of Organic Chemistry. 55 (5): 1649–1664. doi:10.1021 / jo00292a046.
- ^ Hutchings, GJ; Brust, M.; Schmidbaur, H. (2008). “Gold – eine einführende Perspektive”. Bewertungen der Chemical Society. 37 (9): 1759–65. doi:10.1039 / b810747p. PMID 18762825.
- ^ Nijamudheen, A.; Datta, Ayan (2020). “Goldkatalysierte Kreuzkupplungsreaktionen: Ein Überblick über Entwurfsstrategien, mechanistische Studien und Anwendungen”. Chemie: Eine europäische Zeitschrift. 26: 1442–1487. doi:10.1002 / chem.201903377.
- ^ Shen, HC (2008). “Jüngste Fortschritte bei der Synthese von Carbocyclen und Heterocyclen durch homogene Goldkatalyse. Teil 2: Cyclisierungen und Cycloadditionen”. Tetraeder. 64 (34): 7847–7870. doi:10.1016 / j.tet.2008.05.082.
- ^ Reetz, MT; Sommer, K. (2003). “Goldkatalysierte Hydroarylierung von Alkinen”. Europäisches Journal für Organische Chemie. 2003 (18): 3485–3496. doi:10.1002 / ejoc.200300260.
- ^ Nieto-Oberhuber, C.; Muñoz, MP; Buñuel, E.; Nevado, C.; Cárdenas, DJ; Echavarren, AM (2004). “Kationische Gold (I) -Komplexe: Hochalkynophile Katalysatoren für die Exo- und Endocyclisierung von Eninen”. Angewandte Chemie International Edition. 43 (18): 2402–2406. doi:10.1002 / anie.200353207. PMID 15114573.
- ^ Gasparrini, F.; Giovannoli, M.; Misiti, D.; Natile, G.; Palmieri, G.; Maresca, L. (1993). “Gold (III) -katalysierte Eintopfsynthese von Isoxazolen aus terminalen Alkinen und Salpetersäure”. Zeitschrift der American Chemical Society. 115 (10): 4401–4402. doi:10.1021 / ja00063a084.
- ^ Hoffmann-Röder, A.; Krause, N. (2005). “Das goldene Tor zur Katalyse”. Organische und Biomolekulare Chemie. 3 (3): 387–91. doi:10.1039 / b416516k. PMID 15678171.
- ^ Wegner, HA; Auzias, M. (2011). “Gold für CC-Kupplungsreaktionen: ein Schweizer Taschenmesserkatalysator?” Angewandte Chemie International Edition. 50 (36): 8236–47. doi:10.1002 / anie.201101603. PMID 21818831.
- ^ Bandini, Marco (2011-02-01). “Allylalkohole: Nachhaltige Quellen für katalytische enantioselektive Alkylierungsreaktionen”. Angewandte Chemie International Edition. 50 (5): 994–995. doi:10.1002 / anie.201006522. ISSN 1521-3773. PMID 21268189.
- ^ Zhang, Zhibin; Lee, Seong Du; Widenhoefer, Ross A. (2009-04-22). “Intermolekulare Hydroaminierung von Ethylen und 1-Alkenen mit cyclischen Ureas, die durch achirale und chirale Gold (I) -Komplexe katalysiert werden”. Zeitschrift der American Chemical Society. 131 (15): 5372–5373. doi:10.1021 / ja9001162. ISSN 0002-7863. PMC 2891684. PMID 19326908.
- ^ LaLonde, Rebecca L.; Jr., William E. Brenzovich; Benitez, Diego; Tkatchouk, Ekaterina; Kelley, Kotaro; III, William A. Goddard; Toste, F. Dean (2010). “Alkylgoldkomplexe durch intramolekulare Aminoaurierung nicht aktivierter Alkene”. Chemische Wissenschaft. 1 (2): 226. doi:10.1039 / C0SC00255K. PMC 3866133. PMID 24358445.
- ^ Muratore, Michael E.; Homs, Anna; Obradors, Carla; Echavarren, Antonio M. (01.11.2014). “Bewältigung der Herausforderung intermolekularer Gold (I) -katalysierter Cycloadditionen von Alkinen und Allenen”. Chemie: Eine asiatische Zeitschrift. 9 (11): 3066–3082. doi:10.1002 / asia.201402395. ISSN 1861-471X. PMC 4676923. PMID 25048645.
Recent Comments