Dreisträngige DNA – Wikipedia

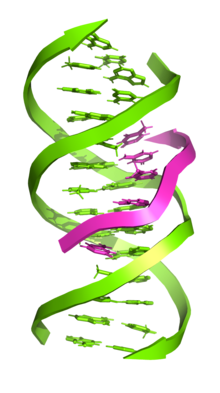
Dreisträngige DNA (auch bekannt als H-DNA oder Triplex-DNA) ist eine DNA-Struktur, in der sich drei Oligonukleotide umeinander wickeln und eine Dreifachhelix bilden. In dreisträngiger DNA bindet der dritte Strang an eine Doppelhelix der B-Form-DNA (über Watson-Crick-Basenpaarung), indem er Hoogsteen-Basenpaare oder umgekehrte Hoogsteen-Wasserstoffbrücken bildet.
Struktur[edit]


Stabilste Dreifachbasenpaarung in dreisträngiger DNA. Rx-Ry: Watson- und Crick-Basenpaarbindung. Ry-Rz: Hoogesteen-Basenpaarbindung.
Hoogsteen Basenpaarung[edit]
Eine Thymin (T) -Nukleobase kann durch Bildung einer Hoogsteen-Wasserstoffbrücke an eine Watson-Crick-Basenpaarung von TA binden. Der Thyminwasserstoff bindet sich an das Adenosin (A) der ursprünglichen doppelsträngigen DNA, um ein TA * T-Basentriplett zu erzeugen.[1] Unter sauren Bedingungen kann ein protoniertes Cytosin, dargestellt als C +, durch Hoogsteen-Basenpaarung ein Basentriplett mit einem CG-Paar bilden und CG * C + bilden. Die TA * T- und CG * C + -Basenpaare sind die am stärksten stabilisierten Triplett-Basenpaare, die sich bilden können, während TA * G und CG * G die am stärksten destabilisierten Triplett-Basenpaare sind.[2]
Intermolekulare und intramolekulare Wechselwirkungen[edit]


Es gibt zwei Klassen von Triplex-DNA: intermolekulare und intramolekulare Formationen. Ein intermolekularer Triplex bezieht sich auf die Triplexbildung zwischen einem Duplex und einem anderen (dritten) DNA-Strang. Der dritte Strang kann entweder von einem benachbarten Chromosom oder einem Triplex-bildenden Oligonukleotid (TFO) stammen. Intramolekulare Triplex-DNA wird aus einem Duplex mit Homopurin- und Homopyrimidinsträngen mit Spiegelwiederholungssymmetrie gebildet.[3] Der Grad der Superwicklung in der DNA beeinflusst das Ausmaß der auftretenden intramolekularen Triplexbildung.[4] Es gibt zwei verschiedene Arten von intramolekularer Triplex-DNA: H-DNA und H * -DNA. Die Bildung von H-DNA wird unter sauren Bedingungen und in Gegenwart zweiwertiger Kationen wie Mg stabilisiert2+. In dieser Konformation biegt sich der Homopyrimidinstrang im Duplex zurück, um parallel an den Purinstrang zu binden. Die zur Stabilisierung dieser Konformation verwendeten Basentriaden sind TA * T und CG * A.+. Das Cytosin dieser Basentriade muss protoniert werden, um diese intramolekulare Dreifachhelix zu bilden, weshalb diese Konformation unter sauren Bedingungen stabilisiert wird.[5] H * -DNA hat günstige Bildungsbedingungen bei neutralem pH und in Gegenwart zweiwertiger Kationen.[4] Diese intramolekulare Konformation entsteht aus der antiparallelen Bindung des Homopurin- und Purinstrangs des Duplex. Es wird durch TA * A- und CG * G-Basentripletts stabilisiert.[3][5]
Funktion[edit]
Triplex bildende Oligonukleotide (TFO)[edit]
TFOs sind kurze (~ 15-25 nt) Nukleinsäurestränge, die in der Hauptfurche doppelsträngiger DNA unter Bildung intramolekularer Triplex-DNA-Strukturen binden. Es gibt Hinweise darauf, dass sie auch die Genaktivität modulieren können in vivo. In der Peptidnukleinsäure (PNA) wird das Zucker-Phosphat-Rückgrat der DNA durch ein proteinartiges Rückgrat ersetzt. PNAs bilden P-Schleifen, während sie mit Duplex-DNA interagieren, und bilden mit einem DNA-Strang einen Triplex, während der andere verdrängt wird. Es wurde angenommen, dass sich unter RecA-Protein im Verlauf der homologen Rekombination sehr ungewöhnliche Rekombinationen oder parallele Triplexe oder R-DNA bilden.[6]
TFOs binden spezifisch an Homopurin-Homopyrimidin-Regionen, die häufig in Promotor- und Intronsequenzen von Genen vorkommen und die Zellsignalisierung beeinflussen.[7] TFOs können die Transkription hemmen, indem sie mit hoher Spezifität an die DNA-Helix binden, wodurch die Bindung und Funktion von Transkriptionsfaktoren für bestimmte Sequenzen blockiert werden. Durch Einführung von TFOs in eine Zelle (durch Transfektion oder andere Mittel) kann die Expression bestimmter Gene gesteuert werden.[8] Diese Anwendung hat neue Auswirkungen auf die ortsspezifische Mutagenese und Gentherapie. In menschlichen Prostatakrebszellen wird ein Transkriptionsfaktor Ets2 überexprimiert, und es wird angenommen, dass er das Wachstum und Überleben von Zellen in einem solchen Überschuss vorantreibt. Carbone et al. entwickelten ein sequenzspezifisches TFO für die Ets2-Promotorsequenz, das die Genexpression herunterregulierte und zu einer Verlangsamung des Zellwachstums und des Zelltods führte.[9] Changxian et al. haben auch einen TFO vorgestellt, der auf die Promotorsequenz von bcl-2 abzielt, einem Gen, das die Apoptose hemmt.[10]
Die beobachtete Hemmung der Transkription kann auch negative Auswirkungen auf die Gesundheit haben, wie z. B. ihre Rolle im rezessiven autosomalen Gen für Friedreichs Ataxie.[11] In Fredricks Ataxie beeinträchtigt die Bildung von Triplex-DNA die Expression von Intron 1 des FXN-Gens. Dies führt zu einer Degeneration des Nervensystems und des Rückenmarks, wodurch die Bewegung der Gliedmaßen beeinträchtigt wird.[12] Um dieser Triplex-Instabilität entgegenzuwirken, wurde gezeigt, dass Nukleotid-Exzisionsreparaturproteine (NERs) dreisträngige DNA-Strukturen erkennen und reparieren, wodurch die vollständige Verfügbarkeit des zuvor inhibierten und instabilen Gens wiederhergestellt wird.[13]


Peptidnukleinsäuren (PNA)[edit]
Peptidnukleinsäuren sind synthetische Oligonukleotide, die dem Protease-Abbau widerstehen und zur Induktion der Reparatur an ortsspezifischen Triplex-Bildungsregionen an genomischen DNA-Stellen verwendet werden. PNAs können durch Watson-Crick-Basenpaarungsbindung mit hoher Affinität und Sequenzspezifität an eine komplementäre DNA-Sequenz binden und durch parallele Orientierung Hoogsteen-Bindungen mit der PNA zum 5′-Ende des DNA-Strangs hin Dreifachhelices bilden.[14] Die PNA-DNA-Triplexe sind stabil, da PNAs aus einem neutral geladenen Pseudopeptidrückgrat bestehen, das an die doppelsträngige DNA (dsDNA) -Sequenz bindet.[15] Ähnlich wie Homopyrimidin in TFOs kann Homopyrimidin in PNAs eine Bindung mit dem komplementären Homopurin in der Zielsequenz der dsDNA bilden. Diese DNA-Analoga können an dsDNA binden, indem sie die Umgebungs-DNA-Bedingungen und verschiedene prädiktive Erkennungsmodi ausnutzen. Dies unterscheidet sich von TFOs, die durch die Hauptrillenerkennung der dsDNA binden[14].
Eine der zur Erkennung verwendeten Vorhersagemodi ist eine Duplexinvasion[16][15]. Innerhalb der gemischten A-T / G-C-dsDNA-Sequenz wird von einem Paar pseudokomplementärer (pc) PNAs angegriffen, die durch doppelte Invasion durch gleichzeitige Bildung von Diaminopurin (D) und Thiouracil (U) an dsDNAs binden könnens) die Adenin bzw. Thymin ersetzen[16]. Das PC-PNA-Paar bildet ein DT und ein U.s -A und GC oder CG Watson-Crick paarten die PNA-DNA-Helix mit jedem der komplementären DNA-Stränge. Eine andere Form der erkannten Duplexinvasion bei der Zielsequenz kann in dsDNA auftreten, die gemischte TC-Sequenzen enthält.[17] Diese Form der Duplexinvasion wird durch eine komplementäre Sequenz von Homopurin-PNA-Oligomeren erreicht. Dieser Triplex wird aus einem PNA-DNA-Hybrid gebildet, der antiparallel zur komplementären DNA-Sequenz bindet und zu einem verdrängten nichtkomplementären DNA-Strang führt.[15]
Zusätzlich kann PNA modifiziert werden, um am Zielort Triplex-Strukturen mit „Klemme“ zu bilden [16] . Eine Art der gebildeten “Klammer” ist eine Bis-PNA-Struktur, in der zwei PNA-Moleküle durch einen flexiblen Linker wie 8-Amino-3,6-dioxaoctansäure (O) zusammengehalten werden.[18] Die Bis-PNA-Struktur bildet einen PNA-DNA-PNA-Triplex an der Zielstelle, wobei ein Strang Watson-Crick-Basenpaare mit DNA in antiparalleler Orientierung bildet und der andere Strang Hoogsteen-Basenpaare mit dem Homopurin-DNA-Strang in der DNA bildet. PNA Duplex.[17] Eine Schwanzklemme PNA (tcPNA) ist auch eine andere Form einer Triplexklemme, die ebenfalls gebildet werden kann. TcPNAs enthalten einen verlängerten Schwanz von 5 bis 10 bp, der zusätzlich zu einer PNA-DNA-PNA- “Klemme” einen PNA / DNA-Duplex bildet. Dies ermöglicht eine spezifischere PNA-Bindung, ohne dass eine Homopyrimidie / Pyridin-Dehnung erforderlich ist.[15] Es wurde gezeigt, dass diese Klemmstrukturen eine hohe Affinität und Spezifität aufweisen. Die Zugabe von Lysinresten an einem oder beiden Enden von PNAs könnte verwendet werden, um die Zellaufnahme und -bindung zu erhöhen[16].
Genetische Regulation[edit]
Dreisträngige DNA war an der Regulation mehrerer Gene beteiligt. Zum Beispiel die c-mein C Das Gen wurde umfassend mutiert, um die Rolle zu untersuchen, die Triplex-DNA im Vergleich zur linearen Sequenz bei der Genregulation spielt. A c-mein C Das Promotorelement, das als Nuklease-sensitives Element oder NSE bezeichnet wird, kann intramolekulare Tandem-Triplexe vom H-DNA-Typ bilden und weist ein repetitives Sequenzmotiv (ACCCTCCCC) auf.4. Das mutierte NSE wurde auf Transkriptionsaktivität und auf seine intra- und intermolekulare Triplexbildungsfähigkeit untersucht. Die Transkriptionsaktivität von mutierten NSEs kann durch die Fähigkeit des Elements zur Bildung von H-DNA vorhergesagt werden und nicht durch die Anzahl, Position oder Anzahl der mutierten Basenpaare. DNA kann daher ein dynamischer Teilnehmer an der Transkription des c-myc-Gens sein.[19]
Genexpression[edit]
Gemäß mehreren veröffentlichten Artikeln hat H-DNA die Fähigkeit, die Genexpression in Abhängigkeit von Faktoren wie Ort und Sequenzen in der Nähe zu regulieren. Obwohl intergene Regionen des prokaryotischen Genoms nur geringe Spuren natürlich vorkommender H-DNA- oder Triplex-Motive aufweisen, haben sich H-DNA-Strukturen im eukaryotischen Genom als häufiger erwiesen. Es wurde gezeigt, dass H-DNA in Säugetierzellen, einschließlich Menschen, besonders häufig vorkommt (1 von 50.000 bp).[14] Genetische Sequenzen, die an der Genregulation beteiligt sind, finden sich typischerweise in den Promotorregionen des eukaryotischen Genoms.[14]
Folglich hat die Promotorregion die Fähigkeit gezeigt, H-DNA mit einer höheren Frequenz zu bilden.[14] Eine bioinformatische Analyse der S. cerevisiae Das Genom beobachtete das Auftreten von H-DNA und anderen dreifachen DNA-Motiven in vier Organisationsregionen: Introns, Exons, Promotorregionen und verschiedenen Regionen. Die Bioinformatik zeigte insgesamt 148 mögliche Strukturen von H-DNA oder Triplett-DNA. Die Promotorregion war für die höhere Frequenz mit 71 Dreifachstrukturen verantwortlich, während die Exons für 57 Dreifachstrukturen und die Introns und Sonstiges für 2 und 18 Strukturen verantwortlich waren.[20]
In-vitro- und In-vivo-Studien zur Expression des eukaryotischen Genoms führten zu einem von drei Ergebnissen: Hochregulierung, Herunterregulierung oder keine Änderung des Vorhandenseins von H-DNA-Motiven.[14] Kato et. al. berichtete Hochregulationsexpression von lacZ, als H-DNA in den B-Lactamase-Promotor eingeführt wurde.[21][14] Andererseits eine ähnliche Studie (Brachmachari et al.) berichteten über keine statistisch signifikante Hemmung der lacZ Reportergen, wenn H-DNA in das Genom von Säuger-COS-Zellen inseriert wurde.[14] Obwohl Studien auf eine Regulation der H-DNA hinweisen, wird der Mechanismus noch untersucht. Potaman et al. assoziiert den Mechanismus der Genregulation mit den Wechselwirkungen zwischen der H-DNA und der TATA-Box in der Promotorregion der Na, K-ATPase. In H-DNA-Formationen neben einer TATA-Box destabilisiert die H-DNA-Struktur die für die Transkription essentiellen TA-Bindungen. Die Interferenz mit der TATA-Box hemmt die Transkriptionsmaschinerie und die Transkriptionsinitiierung, die die Genexpression stören.[14][22] Andere Mechanismen, die mit der genomischen Expression einer genetischen Sequenz in Gegenwart von H-DNA verbunden sind, betreffen TFOs. In-vitro-Studien haben eine Abnahme der Genexpression in Gegenwart von TFOs in Säugetierzellen gezeigt.[23] Ein weiterer möglicher Mechanismus von Valentina et al. legen nahe, dass der 13-mer AG-Motiv-Oligonukleotid-Triplex-Komplex (TFO-Komplex) die Transkription von mRNA durch kompetitive Hemmung herunterreguliert.[24] Die direkte Hemmung der Genexpression aus H-DNA ist der Schlüssel zur Mutagenese, Replikationshemmung und sogar zur DNA-Rekombination im Genom.[14]
Rekombination[edit]


Es wurde gezeigt, dass H-DNA-Motive die homologe Rekombination mit verschiedenen Mechanismen stimulieren. Erste Implikationen für die Rolle von H-DNA bei der Rekombination kamen in den frühen 1990er Jahren bei der Beobachtung von RecA, einem bakteriellen DNA-Rekombinationsprotein, das aus Triple-Helix-DNA besteht. RecA zeigt eine enzymatische Aktivität, die für die Rekombination wesentlich ist.[6][25] Eine homologe Rekombination mit H-DNA-Motiven wurde auch in Eukaryoten gefunden. Es wurde gezeigt, dass RadA, ein zu RecA homologes Protein, bei der Rekombination die gleiche enzymatische Aktivität aufweist wie RecA.[26] Das Protein hat die Fähigkeit, homologe Stränge durch parallele dreisträngige Helices zu fördern und auszutauschen.[27][28] Die einzelsträngige DNA (ssDNA) und die komplementäre doppelsträngige DNA (dsDNA) bilden eine D-Loop-Struktur.[29][14] Ein weiterer möglicher Mechanismus für RecA ist die ssDNA aus zwei getrennten H-DNA-Strukturen zur Bildung von Watson-Crick-Basenpaaren. Die neue Struktur ist als Holliday-Übergang bekannt, ein Zwischenprodukt bei der homologen Rekombination.[14] H-DNA findet sich auch in anderen Formen der Rekombination. In Säugetierzellen zeigten H-DNA-Sequenzen eine hohe Rekombinationsfrequenz. Beispielsweise fand eine an der Myelomzelllinie von Mäusen durchgeführte Studie H-DNA-Strukturen in Cγ2a und Cγ2b, die am Schwesterchromatidaustausch beteiligt sind.[14]
Biologische Implikationen[edit]
Genetische Instabilität[edit]
Beträchtliche Forschungen wurden zu den biologischen Auswirkungen in Bezug auf das Vorhandensein von H-DNA in den Haupt-Breakpoint-Regionen (Mbr) und Doppelstrang-Breakpoints bestimmter Gene durchgeführt. Neuere Arbeiten haben das Vorhandensein von Nicht-B-DNA-Strukturen mit Fällen genetischer Instabilität in Verbindung gebracht.[30]
Polypurin-Spiegel-Repeat-H-DNA-bildende Sequenzen wurden neben dem P1-Promotor des gefunden c-MYC Gen und sind mit den wichtigsten Breakpoint-Hotspots dieser Region verbunden. Fälle von genetischer Instabilität wurden auch bei den F1-Nachkommen transgener Mäuse nach Einbau von humanen H-DNA-bildenden Sequenzen, gepaart mit Z-DNA-Sequenzen, in ihre Genome beobachtet, bei denen zuvor keine Instabilität berichtet wurde.[31] Zusätzlich Bildung von R.RY H-DNA-Konformationen wurden am Mbr des beobachtet bcl-2 Gen. Es wurde vermutet, dass die Bildung dieser Strukturen die bei vielen Krebsarten und den meisten follikulären Lymphomen beobachtete t (14; 18) -Translokation verursacht. Diese Beobachtung hat zu Untersuchungen geführt, die darauf hinwiesen, dass eine erhebliche Abnahme der Translokationsereignisse beobachtet werden kann, nachdem die Bildung von H-DNA durch geringfügige Änderung der Sequenz dieser Region blockiert wurde.[31][32] Es wurde auch beobachtet, dass lange Strecken von GAA · TTC sehr stabile H-DNA-Strukturen bilden. Es wurde gezeigt, dass Wechselwirkungen zwischen diesen beiden H-DNA-Strukturen, die als klebrige DNA bezeichnet werden, die Transkription der DNA unterbrechen X25oder Frataxin-Gen. Da verringerte Spiegel des Proteins Frataxin mit Friedreich-Ataxie assoziiert sind, wurde die Bildung dieser Instabilität als Grundlage für diese genetische Erkrankung vorgeschlagen.[33][34]
Zusätzlich wurde gezeigt, dass H-DNA Mutationen verursacht, die mit kritischen zellulären Prozessen wie DNA-Replikation und -Transkription zusammenhängen.[35] Die Bedeutung dieser Prozesse für das Überleben hat zur Entwicklung komplexer DNA-Reparaturmechanismen geführt, mit denen Zellen DNA-Schäden erkennen und beheben können. Nicht-kanonische DNA-Strukturen können als Schädigung durch die Zelle wahrgenommen werden, und neuere Arbeiten haben eine erhöhte Prävalenz von Mutationen in der Nähe von nicht-B-DNA-bildenden Sequenzen gezeigt.[35] Einige dieser Mutationen sind auf die Wechselwirkungen zwischen H-DNA und den an der DNA-Replikation und -Transkription beteiligten Enzymen zurückzuführen, wobei H-DNA diese Prozesse stört und verschiedene DNA-Reparaturmechanismen auslöst. Dies kann zu genetischer Instabilität führen und impliziert H-DNA bei der Krebsentstehung.[35]
DNA Replikation[edit]
Es wurde gezeigt, dass die DNA-Replikation die Funktion verschiedener DNA-Reparaturenzyme beeinflusst. Die Bildung von H-DNA beinhaltet die Bildung von einzelsträngiger DNA (ssDNA), die anfälliger für Angriffe durch Nukleasen ist.[35] Es wurde gezeigt, dass verschiedene Nukleasen replikationsabhängig oder replikationsunabhängig mit H-DNA interagieren.[35]
Eine Studie mit menschlichen Zellen ergab, dass die Nucleotide Excision Repair (NER) -Nukleasen ERCC1-XPF und ERCC1-XPG eine genetische Instabilität induzierten.[36] Diese Enzyme spalten H-DNA an der Schleife, die von den beiden wasserstoffgebundenen Strängen von Hoogsteen und dem 5′-Ende des anderen wasserstoffgebundenen Strangs von Watson-Crick gebildet wird.[36] Es wurde gezeigt, dass diese Spaltung große Deletionen induziert, die Doppelstrangbrüche (DSBs) in der DNA verursachen, die zu genetischer Instabilität führen können.[35][36] In Zellen, denen ERCC1-XPF und ERCC1-XPG fehlen, waren diese Deletionen in der Nähe von H-DNA-bildenden Sequenzen weniger häufig.[36] Zusätzlich wurden mehr Mutationen in ERCC1-XPF- und ERCC1-XPG-defizienten Zellen in Abwesenheit einer DNA-Replikation gefunden, was darauf hindeutet, dass sie H-DNA auf replikationsunabhängige Weise verarbeiten.[36]
Alternativ wurde gefunden, dass die DNA-Replikationsreparaturnuklease FEN1 die genetische Instabilität unterdrückt.[36] Ähnlich wie ERCC1-XPG spaltet FEN1 H-DNA am 5′-Ende des Strangs, der nicht an der Hoogsteen-Wasserstoffbindung beteiligt ist.[36]HeLa-Zellen, denen FEN1 fehlt, zeigten eine höhere Prävalenz von Deletionen in der Nähe von H-DNA-bildenden Sequenzen, aber die H-DNA-induzierte Mutagenese war in FEN1-defizienten Zellen in Gegenwart einer DNA-Replikation stärker ausgeprägt.[36] Dies legt nahe, dass FEN1 die H-DNA-induzierte Mutagenese in replikationsabhängiger Weise unterdrückt.[36]
H-DNA wurde aufgrund der Prävalenz von H-DNA-bildenden Sequenzen in der Nähe von Translokationsbruchpunkten in Krebsgenomen in die Ätiologie des menschlichen Krebses einbezogen.[36] Replikationsvermittelte Nukleaseaktivität mit H-DNA zeigt einen anderen Weg der H-DNA-induzierten Mutagenese auf und führt zu Krebswachstum.
Transkription[edit]
H-DNA-bildende Sequenzen können auch genetische Instabilität verursachen, indem sie die Transkription stören und vorzeitig stoppen.[35] Das an der Transkription beteiligte DNA-Abwickeln macht es anfälliger für Schäden. Bei der transkriptionsgekoppelten Reparatur (TCR) stoppt eine Läsion am Matrizenstrang der DNA die Funktion der RNA-Polymerase und signalisiert TCR-Faktoren, um den Schaden durch Ausschneiden zu beheben.[37] H-DNA kann als eine dieser Läsionen wahrgenommen werden.
Eine Studie zur Beobachtung der Transkription durch T7-RNA-Polymerase an einem stabilen H-DNA-bildenden Sequenzanalogon ergab eine Transkriptionsblockade an der Duplex-Triplex-Verbindung. Hier war der Matrizenstrang der zentrale Strang der H-DNA, und die Schwierigkeit, seine Watson-Crick- und Hoogsteen-Wasserstoffbrücken zu zerstören, verhinderte das Fortschreiten der Transkription.[38]
Bei der Transkription durch T7 wurde auf dem P0-Promotor des c-MYC Gen, zeigten die verkürzten Transkriptionsprodukte, die gefunden wurden, dass die Transkription in unmittelbarer Nähe der H-DNA-Bildungssequenz stromabwärts des Promotors gestoppt wurde. Die Bildung von H-DNA in dieser Region verhindert, dass T7 aufgrund der dadurch verursachten sterischen Hinderung den Matrizenstrang hinunter wandert. Dies stoppt die Transkription und signalisiert, dass TCR-Faktoren die H-DNA auflösen, was zu einer DNA-Exzision führt, die eine genetische Instabilität verursachen kann.[37] Die Spiegelsymmetrie und Prävalenz von Guaninresten in der c-MYC Gen gibt es eine hohe Neigung zur nicht-kanonischen DNA-Strukturbildung.[39] Dies zusammen mit der Aktivität von TCR-Faktoren während der Transkription macht es stark mutagen, wobei es eine Rolle bei der Entwicklung von Burkitt-Lymphom und Leukämie spielt.[37][39]
Anwendungen[edit]
Die dreisträngigen DNA-Regionen können durch die Assoziation von Triplex-bildenden Oligonukleotiden (TFO) und Peptidnukleinsäuren (PNAs) erzeugt werden. In der Vergangenheit wurde gezeigt, dass die TFO-Bindung die Transkription, Replikation und Proteinbindung an DNA hemmt.[16] Es wurde auch gezeigt, dass an Mutagene gebundene TFOs DNA-Schäden fördern und Mutagenese induzieren [14]. Obwohl bekannt ist, dass TFO die Transkription und Replikation von DNA behindert, haben neuere Studien gezeigt, dass TFO verwendet werden kann, um ortsspezifische Genmodifikationen sowohl in vitro als auch in vivo zu vermitteln[16]. Eine andere kürzlich durchgeführte Studie hat auch gezeigt, dass TFOs zur Unterdrückung von Onkogenen und Protoonkogenen verwendet werden können, um das Wachstum von Krebszellen zu reduzieren. Beispielsweise hat eine kürzlich durchgeführte Studie TFOs verwendet, um den Zelltod in Hepatomzellen durch die Verringerung der MET-Expression zu verringern.
PNA-TFOs können die Rekombinationsfrequenzen verbessern, was zu einer gezielten, spezifischen Bearbeitung von Genen führt. Die PNA-DNA-PNA-Triplex-Helix kann durch den zelleigenen DNA-Reparaturmechanismus erkannt werden, der die umgebende DNA für die homologe Rekombination sensibilisiert. Damit eine ortsspezifische PNA-Struktur die Rekombination innerhalb einer DNA-Sequenz vermittelt, kann eine Bis-PNA-Struktur mit einem 40-nt-DNA-Fragment gekoppelt werden, das zu einer benachbarten Region auf dem Zielgen homolog ist.[17] Es wurde gezeigt, dass die Verknüpfung eines TFO mit einem Donor-DNA-Strang die Rekombination des Zielgens und der angrenzenden Genzielregion induziert[40]. Der Mechanismus für diese Form der Rekombination und Reparatur wurde mit dem NER-Weg (Nucleotide Excision Repair) verknüpft, der eine Rolle bei der Erkennung und Reparatur von Triplex-Strukturen spielt.[17][16] Mehrere Untersuchungen legen nahe, dass die Xeroderma pigmentosum Gruppe A (XPA) und das Replikationsprotein A (RPA), die NER-Faktoren sind, spezifisch als Komplex an vernetzte Triplexstrukturen binden können. Es ist bekannt, dass dieser Mechanismus neben anderen eine Rolle bei der Erkennung und Reparatur von Triplex-Strukturen spielt.
Die In-vivo-Abgabe von TFOs war ein Haupthindernis bei der Verwendung von TFOs zur Genmodifikation.[41] Eine Studie zum In-vivo-Targeting von hämatopoetischen Stammzellen schlug eine neuartige Technik zur Konjugation von PNA-Molekülen mit zellpenetrierendem Peptid (CPPs) neben Poly (milch-co-glykolsäure) (PLGA) -Nanopartikeln vor, um 6-bp-Modifikationen im CCR5-Gen zu ermöglichen.[40] Die Bearbeitung des CCR5-Gens wurde mit der HIV-1-Resistenz in Verbindung gebracht.[42] CPPs sind Proteine, die in der Lage sind, „Fracht“ wie kleine Proteine oder Moleküle erfolgreich in Zellen zu transportieren. Die PGLAs sind biologisch abbaubares Material, das PNA-Moleküle als Nanopartikel für ortsspezifische Genommodifikationen einkapselt.[40] Die Studie ergab, dass die PNA-DNA-PGLA-Nanopartikel die hämatopoetischen Stammzellen mit geringerer Toxizität und virusfrei effektiv bearbeiten konnten und die Konjugation mit CPP ein direktes Targeting der Gene für die ortsspezifische Mutagenese in den Stammzellen ermöglichte.
In einer neuartigen Studie zur Gentherapie bei Mukoviszidose (CF) wurden drei Schwanzklemmpeptidnukleinsäuren (PNAs) neben dem Donor-DNA-Molekül so konstruiert, dass sie von Nanopartikeln abgegeben werden, um F508 del-Mutationen auf dem Transmembran-Leitfähigkeitsregler für Mukoviszidose (CFTR) in zu korrigieren humane Bronchialepithelzellen in vivo und in vitro.[43] Die F508 del-Mutation ist die am häufigsten auftretende Mutation, die dazu führt, dass eine Person an CF leidet.[44] Die F508-Mutation führt zu einem Funktionsverlust der CFTR, einem Plasmamembranchloridkanal, der durch ein cyclisches Adenosinmonophosphat (cAMP) reguliert wird. In dieser Studie konnten sie den neuartigen Behandlungsansatz für CF mithilfe von Nanopartikeln zur Korrektur des F508 del entwickeln CFTR Mutation sowohl in vitro in humanen Bronchialepithelzellen (HBE) als auch in vivo in einem CF-Mausmodell, die zum Auftreten eines CFTR-abhängigen Chloridtransports führte.[43]
Geschichte[edit]


Dreisträngige DNA-Strukturen waren in den 1950er Jahren gängige Hypothesen, als Wissenschaftler Schwierigkeiten hatten, die wahre Strukturform der DNA zu entdecken. Watson und Crick (die später den Nobelpreis für ihr Doppelhelixmodell erhielten) betrachteten ursprünglich ein Dreifachhelixmodell, ebenso wie Pauling und Corey, die 1953 einen Vorschlag für ihr Dreifachhelixmodell veröffentlichten.[45][46] sowie Mitwissenschaftler Fraser.[47] Watson und Crick identifizierten jedoch bald mehrere Probleme mit diesen Modellen:
- Negativ geladene Phosphate in der Nähe der Achse stoßen sich gegenseitig ab und lassen die Frage offen, wie die Dreikettenstruktur zusammen bleibt.
- In einem Triple-Helix-Modell (insbesondere dem Modell von Pauling und Corey) scheinen einige der Van-der-Waals-Abstände zu klein zu sein.
Frasers Modell unterschied sich von Pauling und Coreys darin, dass sich in seinem Modell die Phosphate außen und die Basen innen befinden und durch Wasserstoffbrücken miteinander verbunden sind. Watson und Crick fanden jedoch Frasers Modell zu schlecht definiert, um seine Unzulänglichkeiten speziell zu kommentieren.
Eine alternative dreisträngige DNA-Struktur wurde 1957 beschrieben.[48] Felsenfeld, Davies und Rich sagten voraus, dass, wenn ein Strang nur Purine und der andere Strang nur Purine enthielt, der Strang eine Konformationsänderung erfahren würde, um eine dreisträngige DNA-Helix zu bilden. Es wurde vorausgesagt, dass die dreisträngige DNA (H-DNA) aus einem Polypurin- und zwei Polypyrimidinsträngen besteht.[6][48] Es wurde angenommen, dass es nur in einem biologischen In-vivo-Prozess auftritt: als Zwischenprodukt während der Wirkung des E. coli-Rekombinationsenzyms RecA.[48] Frühe Modelle in den 1960er Jahren sagten die Bildung von Komplexen zwischen Polycetiyl- und Guanin-Oligonukleotiden voraus. Die Modelle schlugen Wechselwirkungen vor, die als Hoogsten-Paarung (Nicht-Watson-Crick-Wechselwirkungen) in der Hauptrille bekannt sind.[6] Kurz darauf wurden Dreifachhelices aus einem Pyrimidin- und zwei Purinsträngen vorhergesagt.[6] Die Entdeckung von in H-DNA-Abschnitten in supergewickelten Plasmiden weckte das moderne Interesse an der möglichen Funktion von Triplex-Strukturen in lebenden Zellen.[49] Zusätzlich wurde bald gefunden, dass Homopyrimidin und einige Purin-reiche Oligonukleotide eine stabile H-DNA-Struktur mit den Homopurin-Homopyrimidin-Bindungssequenz-spezifischen Strukturen auf den DNA-Doppelsträngen bilden können.[50]
Verweise[edit]
- ^ Rhee S., Han Z., Liu K., Miles HT, Davies DR (Dezember 1999). “Struktur einer dreifach helikalen DNA mit einem Triplex-Duplex-Übergang”. Biochemie. 38 (51): 16810–5. doi:10.1021 / bi991811m. PMID 10606513.
- ^ Mergny JL, Sun JS, Rougée M., Montenay-Garestier T., Barcelo F., Chomilier J., Hélène C. (Oktober 1991). “Sequenzspezifität bei der Dreifachhelixbildung: experimentelle und theoretische Untersuchungen zum Einfluss von Fehlpaarungen auf die Triplexstabilität”. Biochemie. 30 (40): 9791–8. doi:10.1021 / bi00104a031. PMID 1911764.
- ^ ein b Ussery DW, Sinden RR (Juni 1993). “Umwelteinflüsse auf das In-vivo-Niveau der intramolekularen Triplex-DNA in Escherichia coli”. Biochemie. 32 (24): 6206–13. doi:10.1021 / bi00075a013. PMID 8512930.
- ^ ein b Dayn A, Samadashwily GM, Mirkin SM (Dezember 1992). “Intramolekulare DNA-Triplexe: ungewöhnliche Sequenzanforderungen und Einfluss auf die DNA-Polymerisation”. Verfahren der National Academy of Sciences der Vereinigten Staaten von Amerika. 89 (23): 11406–10. Bibcode:1992PNAS … 8911406D. doi:10.1073 / pnas.89.23.11406. PMC 50559. PMID 1454828.
- ^ ein b Lyamichev VI, Mirkin SM, Dr. Frank-Kamenetskii (Februar 1986). “Strukturen des Homopurin-Homopyrimidin-Trakts in superhelikaler DNA”. Journal of Biomolecular Structure & Dynamics. 3 (4): 667–9. doi:10.1080 / 07391102.1986.10508454. PMID 3271043.
- ^ ein b c d e Frank-Kamenetskii, MD, Mirkin SM (1995-01-01). “Triplex-DNA-Strukturen”. Jahresrückblick Biochemie. 64: 65–95. doi:10.1146 / annurev.bi.64.070195.000433. PMID 7574496. S2CID 21426188.
- ^ Brázdová M., Tichý V., Helma R., Bažantová P., Polášková A., Krejčí A. et al. (2016). “p53 bindet spezifisch Triplex-DNA in vitro und in Zellen”. Plus eins. 11 (12): e0167439. Bibcode:2016PLoSO..1167439B. doi:10.1371 / journal.pone.0167439. PMC 5131957. PMID 27907175.
- ^ Graham MK, Brown TR, Miller PS (April 2015). “Targeting des menschlichen Androgenrezeptor-Gens mit platinierten triplexbildenden Oligonukleotiden”. Biochemie. 54 (13): 2270–82. doi:10.1021 / bi501565n. PMID 25768916.
- ^ Carbone GM, Napoli S., Valentini A., Cavalli F., Watson DK, Catapano CV (03.08.2004). “Triplex-DNA-vermittelte Herunterregulierung der Ets2-Expression führt zu Wachstumshemmung und Apoptose in menschlichen Prostatakrebszellen.”. Nukleinsäureforschung. 32 (14): 4358–67. doi:10.1093 / nar / gkh744. PMC 514370. PMID 15314206.
- ^ Shen C., Rattat D., Buck A., Mehrke G., Polat B., Ribbert H. et al. (Februar 2003). “Targeting von bcl-2 durch triplexbildendes Oligonukleotid – ein vielversprechender Träger für die Gen-Strahlentherapie”. Krebs-Biotherapie & Radiopharmazeutika. 18 (1): 17–26. doi:10.1089 / 108497803321269296. PMID 12667305.
- ^ Sakamoto N., Chastain PD, Parniewski P., Ohshima K., Pandolfo M., Griffith J. D., Wells RD (April 1999). “Sticky DNA: Selbstassoziationseigenschaften langer GAA.TTC-Wiederholungen in RRY-Triplex-Strukturen aus Friedreichs Ataxie”. Molekulare Zelle. 3 (4): 465–75. doi:10.1016 / s1097-2765 (00) 80474-8. PMID 10230399.
- ^ Bacolla A, Wells RD (April 2009). “Nicht-B-DNA-Konformationen als Determinanten von Mutagenese und menschlicher Krankheit”. Molekulare Karzinogenese. 48 (4): 273–85. doi:10.1002 / mc.20507. PMID 19306308. S2CID 5493647.
- ^ Kaushik Tiwari M., Adaku N., Peart N., Rogers FA (September 2016). “Triplex-Strukturen induzieren DNA-Doppelstrangbrüche durch Kollaps der Replikationsgabel in NER-defizienten Zellen.”. Nukleinsäureforschung. 44 (16): 7742–54. doi:10.1093 / nar / gkw515. PMC 5027492. PMID 27298253.
- ^ ein b c d e f G h ich j k l m n Jain A, Wang G, Vasquez KM (August 2008). “DNA-Dreifachhelices: biologische Konsequenzen und therapeutisches Potenzial”. Biochimie. 90 (8): 1117–30. doi:10.1016 / j.biochi.2008.02.011. PMC 2586808. PMID 18331847.
- ^ ein b c d Hansen ME, Bentin T., Nielsen PE (Juli 2009). Triplex-Targeting von doppelsträngiger DNA mit hoher Affinität unter Verwendung chemisch modifizierter Peptidnukleinsäureoligomere. Nukleinsäureforschung. 37 (13): 4498–507. doi:10.1093 / nar / gkp437. PMC 2715256. PMID 19474349.
- ^ ein b c d e f G Ricciardi AS, McNeer NA, Anandalingam KK, Saltzman WM, Glazer PM (2014). Wajapeyee N (Hrsg.). “Gezielte Genommodifikation durch Dreifachhelixbildung”. Methoden der Molekularbiologie. New York, NY: Springer New York. 1176: 89–106. doi:10.1007 / 978-1-4939-0992-6_8. ISBN 978-1-4939-0991-9. PMC 5111905. PMID 25030921.
- ^ ein b c d Rogers FA, Vasquez KM, Egholm M., Glazer PM (Dezember 2002). Ortsgerichtete Rekombination über bifunktionelle PNA-DNA-Konjugate. Verfahren der National Academy of Sciences der Vereinigten Staaten von Amerika. 99 (26): 16695–700. doi:10.1073 / pnas.262556899. PMC 139206. PMID 12461167.
- ^ Montazersaheb S., Hejazi MS, Nozad Charoudeh H. (November 2018). “Potenzial von Peptidnukleinsäuren in zukünftigen therapeutischen Anwendungen”. Advanced Pharmaceutical Bulletin. 8 (4): 551–563. doi:10.15171 / apb.2018.064. PMC 6311635. PMID 30607328.
- ^ Firulli AB, Maibenco DC, Kinniburgh AJ (April 1994). “Die Triplexbildungsfähigkeit eines c-myc-Promotorelements sagt die Promotorstärke voraus”. Archiv für Biochemie und Biophysik. 310 (1): 236–42. doi:10.1006 / abbi.1994.1162. PMID 8161210.
- ^ Zain R, Sun JS (Mai 2003). “Treten natürliche DNA-Dreifachhelixstrukturen in vivo auf und funktionieren sie?” Zelluläre und molekulare Biowissenschaften. 60 (5): 862–70. doi:10.1007 / s00018-003-3046-3. PMID 12827276.
- ^ Kato M, Shimizu N (Oktober 1992). “Wirkung der potentiellen Triplex-DNA-Region auf die In-vitro-Expression des bakteriellen Beta-Lactamase-Gens in superhelikalen rekombinanten Plasmiden”. Journal of Biochemistry. 112 (4): 492–4. doi:10.1093 / oxfordjournals.jbchem.a123927. PMID 1491004.
- ^ Potaman VN, Ussery DW, Sinden RR (Juni 1996). “Bildung einer kombinierten H-DNA / offenen TATA-Box-Struktur in der Promotorsequenz des menschlichen Na, K-ATPase alpha2-Gens”. Das Journal of Biological Chemistry. 271 (23): 13441–7. doi:10.1074 / jbc.271.23.13441. PMID 8662935.
- ^ Seidman MM, Glazer PM (August 2003). “Das Potenzial zur Genreparatur durch Dreifachhelixbildung”. Das Journal of Clinical Investigation. 112 (4): 487–94. doi:10.1172 / JCI19552. PMC 171401. PMID 12925687.
- ^ Rapozzi V., Cogoi S., Spessotto P., Risso A., Bonora GM, Quadrifoglio F., Xodo LE (Januar 2002). “Antigeneffekt in K562-Zellen eines PEG-konjugierten triplexbildenden Oligonukleotids, das auf das bcr / abl-Onkogen abzielt”. Biochemie. 41 (2): 502–10. doi:10.1021 / bi011314h. PMID 11781088.
- ^ Bertucat G., Lavery R., Prévost C. (Dezember 1998). “Ein Modell für die parallele Dreifachhelixbildung durch RecA: Einzel-Einzel-Assoziation mit einem homologen Duplex über die Nebenrille”. Journal of Biomolecular Structure & Dynamics. 16 (3): 535–46. doi:10.1080 / 07391102.1998.10508268. PMID 10052612.
- ^ Chen J., Tang Q, Guo S., Lu C., Le S., Yan J. (September 2017). Parallele Triplexstruktur zwischen gestreckter einzelsträngiger DNA und homologer Duplex-DNA. Nukleinsäureforschung. 45 (17): 10032–10041. doi:10.1093 / nar / gkx628. PMC 5622322. PMID 28973442.
- ^ Camerini-Otero RD, Hsieh P. (April 1993). “Parallele DNA-Triplexe, homologe Rekombination und andere homologieabhängige DNA-Wechselwirkungen”. Zelle. 73 (2): 217–23. doi:10.1016 / 0092-8674 (93) 90224-e. PMID 8477443.
- ^ Bertucat G., Lavery R., Prévost C. (September 1999). “Ein molekulares Modell für den RecA-geförderten Strangaustausch über parallele dreisträngige Helices”. Biophysical Journal. 77 (3): 1562–76. doi:10.1016 / S0006-3495 (99) 77004-9. PMID 10465767.
- ^ Yang H., Zhou C., Dhar A., Pavletich NP (Oktober 2020). “Mechanismus des Strangaustauschs aus synaptischen RecA-DNA- und D-Loop-Strukturen”. Natur. 586 (7831): 801–806. doi:10.1038 / s41586-020-2820-9. PMID 33057191.
- ^ McKinney JA, Wang G., Mukherjee A., Christensen L., Subramanian SH, Zhao J., Vasquez KM (Januar 2020). “Unterschiedliche DNA-Reparaturwege verursachen genomische Instabilität bei alternativen DNA-Strukturen.”. Naturkommunikation. 11 (1): 236. Bibcode:2020NatCo..11..236M. doi:10.1038 / s41467-019-13878-9. PMC 6957503. PMID 31932649.
- ^ ein b Wang G, Vasquez KM (Juli 2014). “Einfluss alternativer DNA-Strukturen auf DNA-Schäden, DNA-Reparatur und genetische Instabilität”. DNA-Reparatur. 19: 143–51. doi:10.1016 / j.dnarep.2014.03.017. PMC 4216180. PMID 24767258.
- ^ Raghavan SC, Chastain P., Lee J. S., Hegde BG, Houston S., Langen R., et al. (Juni 2005). “Hinweise auf eine Triplex-DNA-Konformation in der bcl-2-Hauptbruchpunktregion der t (14; 18) -Translokation”. Das Journal of Biological Chemistry. 280 (24): 22749–60. doi:10.1074 / jbc.M502952200. PMID 15840562.
- ^ Wang G, Vasquez KM (September 2004). “Natürlich vorkommende H-DNA-bildende Sequenzen sind in Säugetierzellen mutagen.”. Verfahren der National Academy of Sciences der Vereinigten Staaten von Amerika. 101 (37): 13448–53. Bibcode:2004PNAS..10113448W. doi:10.1073 / pnas.0405116101. PMC 518777. PMID 15342911.
- ^ Vetcher AA, Napierala M, Iyer RR, Chastain PD, Griffith JD, Wells RD (Oktober 2002). “Sticky DNA, ein langer GAA.GAA.TTC-Triplex, der intramolekular in der Sequenz von Intron 1 des Frataxin-Gens gebildet wird”. Das Journal of Biological Chemistry. 277 (42): 39217–27. doi:10.1074 / jbc.M205209200. PMID 12161437.
- ^ ein b c d e f G Wang G, Vasquez KM (Januar 2017). “Auswirkungen der Replikation und Transkription auf die DNA-Struktur-bezogene genetische Instabilität”. Gene. 8 (1): 17. doi:10.3390 / gene8010017. PMC 5295012. PMID 28067787.
- ^ ein b c d e f G h ich j Zhao J., Wang G., Del Mundo IM, McKinney JA, Lu X, Bacolla A. et al. (Januar 2018). “Unterschiedliche Mechanismen der Nuklease-gerichteten DNA-Struktur-induzierten genetischen Instabilität in Krebsgenomen”. Zellenberichte. 22 (5): 1200–1210. doi:10.1016 / j.celrep.2018.01.014. PMC 6011834. PMID 29386108.
- ^ ein b c Belotserkovskii BP, De Silva E., Tornaletti S., Wang G., Vasquez KM, Hanawalt PC (November 2007). “Eine triplexbildende Sequenz aus dem menschlichen c-MYC-Promotor stört die DNA-Transkription.”. Das Journal of Biological Chemistry. 282 (44): 32433–41. doi:10.1074 / jbc.M704618200. PMID 17785457. S2CID 24211097.
- ^ Pandey S., Ogloblina AM, Belotserkovskii BP, Dolinnaya NG, Yakubovskaya MG, Mirkin SM, Hanawalt PC (August 2015). Transkriptionsblockade durch stabile H-DNA-Analoga in vitro. Nukleinsäureforschung. 43 (14): 6994–7004. doi:10.1093 / nar / gkv622. PMC 4538819. PMID 26101261.
- ^ ein b
- ^ ein b c McNeer NA, Schleifman EB, Cuthbert A., Brehm M., Jackson A., Cheng C. et al. (Juni 2013). “Die systemische Abgabe von triplexbildender PNA und Donor-DNA durch Nanopartikel vermittelt die ortsspezifische Genombearbeitung menschlicher hämatopoetischer Zellen in vivo.”. Gentherapie. 20 (6): 658–69. doi:10.1038 / gt.2012.82. PMC 3713483. PMID 23076379.
- ^ Hnedzko D, Cheruiyot SK, Rozners E (September 2014). “Verwendung von dreifach helixbildenden Peptidnukleinsäuren zur sequenzselektiven Erkennung doppelsträngiger RNA”. Aktuelle Protokolle in der Nukleinsäurechemie. 58: 4.60.1–23. doi:10.1002 / 0471142700.nc0460s58. ISSN 1934-9270. PMC 4174339. PMID 25199637.
- ^ Schleifman EB, Bindra R., Leif J., del Campo J., Rogers FA, Uchil P. et al. (September 2011). “Gezielte Störung des CCR5-Gens in menschlichen hämatopoetischen Stammzellen, die durch Peptidnukleinsäuren stimuliert werden”. Chemie & Biologie. 18 (9): 1189–98. doi:10.1016 / j.chembiol.2011.07.010. PMC 3183429. PMID 21944757.
- ^ ein b McNeer NA, Anandalingam K., Fields RJ, Caputo C., Kopic S., Gupta A. et al. (April 2015). “Nanopartikel, die triplexbildende Peptidnukleinsäuremoleküle liefern, korrigieren die F508del-CFTR im Atemwegsepithel.”. Naturkommunikation. 6 (1): 6952. doi:10.1038 / ncomms7952. PMC 4480796. PMID 25914116.
- ^ Lukacs GL, Verkman AS (Februar 2012). CFTR: Falten, Fehlfalten und Korrigieren des ΔF508-Konformationsfehlers. Trends in der molekularen Medizin. 18 (2): 81–91. doi:10.1016 / j.molmed.2011.10.003. PMC 3643519. PMID 22138491.
- ^ Pauling L, Corey RB (Februar 1953). “Eine vorgeschlagene Struktur für die Nukleinsäuren”. Verfahren der National Academy of Sciences der Vereinigten Staaten von Amerika. 39 (2): 84–97. Bibcode:1953PNAS … 39 … 84P. doi:10.1073 / pnas.39.2.84. PMC 1063734. PMID 16578429.
- ^ Pauling L, Corey RB (Februar 1953). “Struktur der Nukleinsäuren”. Natur. 171 (4347): 346. Bibcode:1953Natur.171..346P. doi:10.1038 / 171346a0. PMID 13036888. S2CID 4151877.
- ^ Fraser RD (März 2004). “Die Struktur der Desoxyribose-Nukleinsäure”. Zeitschrift für Strukturbiologie. 145 (3): 184–5. doi:10.1016 / j.jsb.2004.01.001. PMID 14997898.
- ^ ein b c Felsenfeld G, Davies DR, Rich A (April 1957). “Bildung eines dreisträngigen Polynukleotidmoleküls”. Zeitschrift der American Chemical Society. 79 (8): 2023–4. doi:10.1021 / ja01565a074.
- ^ Hanvey JC, Shimizu M., Wells RD (September 1988). “Intramolekulare DNA-Triplexe in supergewickelten Plasmiden”. Verfahren der National Academy of Sciences der Vereinigten Staaten von Amerika. 85 (17): 6292–6. doi:10.1073 / pnas.85.17.6292. PMC 281955. PMID 3413097.
- ^ Mirkin SM, Lyamichev VI, Drushlyak KN, Dobrynin VN, Filippov SA, Dr. Frank-Kamenetskii (Dezember 1987). “Die DNA H-Form erfordert eine Homopurin-Homopyrimidin-Spiegelwiederholung”. Natur. 330 (6147): 495–7. doi:10.1038 / 330495a0. PMID 2825028.
Weiterführende Literatur[edit]
Recent Comments