Tình trạng này được di truyền theo cách thống trị tự phát
Đặc biệt
Bệnh thấp khớp [1] Ngoài ra, những người mắc SCS nặng hơn có thể bị tâm thần nhẹ đến trung bình chậm phát triển hoặc khuyết tật học tập. Tùy thuộc vào mức độ nghiêm trọng, một số người mắc SCS có thể yêu cầu một số hình thức can thiệp y tế hoặc phẫu thuật. [2] Hầu hết những người bị SCS sống cuộc sống khá bình thường, bất kể có cần điều trị y tế hay không. [1]
Dấu hiệu và triệu chứng [19659010] [ chỉnh sửa ]
Các cá nhân với SCS đều bị ảnh hưởng khác nhau. Ngay cả trong cùng một gia đình, các cá nhân bị ảnh hưởng có các tính năng khác nhau. Phần lớn các cá nhân mắc SCS bị ảnh hưởng vừa phải, với các đặc điểm trên khuôn mặt không đồng đều và khuôn mặt tương đối phẳng do hốc mắt kém phát triển, xương gò má và hàm dưới. Ngoài những bất thường về thể chất, những người mắc SCS cũng gặp phải sự chậm trễ tăng trưởng, dẫn đến tầm vóc tương đối ngắn. Mặc dù, hầu hết các cá nhân mắc SCS đều có trí thông minh bình thường, một số cá nhân có thể bị chậm phát triển tâm thần từ nhẹ đến trung bình (IQ từ 50 sắt70). Các trường hợp nghiêm trọng hơn của SCS, với biến dạng khuôn mặt nghiêm trọng hơn, xảy ra khi nhiều vết khâu sọ bị đóng sớm. [1]
Khiếm khuyết sọ [ chỉnh sửa ]
Bàn tay của một bệnh nhân có nhiều chữ số
Đầu và mặt phẳng, không đối xứng [2]
Đầu thường có hình nón (acrocephaly) hoặc phẳng (brachycephaly) nhưng cũng có thể dài và hẹp (dolichocephaly) [3]
Đầu ngắn từ trước ra sau [3]
Khuôn mặt bị lệch [3]
Đường chân tóc thấp khiến trán trông cao và rộng [4]
Khiếm khuyết của bàn tay và bàn chân [ chỉnh sửa ]
Khiếm khuyết ở mắt ]
Một sơ đồ cho thấy một khe hở môi và vòm miệng hoàn toàn
Khiếm khuyết tai, mũi và miệng [ chỉnh sửa ]
Tai nhỏ, thấp có thể là xoay một chút về phía sau và có một pinna (phồng) nổi bật [3]
Mũi bị cong (hơi cong xuống ở đỉnh) hơi lệch khỏi trung tâm một d chứa một vách ngăn lệch [1]
Malocclusion liên quan đến các bất thường về răng bao gồm giảm men răng (men răng mỏng do hình thành không hoàn chỉnh), hyperdontia (răng thừa) và răng hàm (răng nhỏ, hình dạng bất thường) [5]
19659031] Các khuyết tật ít phổ biến hơn [ chỉnh sửa ]
Craniosynostosis [ chỉnh sửa ]
Chỉ khâu sọ được nhìn từ đỉnh đầu
ba phần chính bao gồm nền tảng của cranium (xương chẩm), mặt (xương trán) và đỉnh (xương đỉnh) và hai bên (xương thái dương) của đầu. Hầu hết các xương của cranium được đặt vĩnh viễn vào vị trí trước khi sinh. Tuy nhiên, xương thái dương và xương được phân tách bằng chỉ khâu, vẫn mở, cho phép đầu hơi thay đổi hình dạng trong khi sinh. Các chỉ khâu sọ cuối cùng đóng lại trong vài năm đầu tiên sau khi sinh, sau khi não đã phát triển xong. [1]
Ở những người bị SCS, chỉ khâu mạch vành tách xương trước ra khỏi xương paralal, đôi khi còn đóng lại sớm (craniosynostosis) Sinh. Nếu chỉ khâu mạch vành đóng không đối xứng hoặc đơn phương, thì mặt và trán sẽ hình thành không đều nhau, từ bên này sang bên kia. Những người bị SCS có đầu nhọn, giống như tháp vì não của họ phát triển nhanh hơn hộp sọ, dẫn đến tăng áp lực nội sọ (ICP) và khiến đỉnh đầu và / hoặc trán phình ra để cho phép não phát triển. Khuôn mặt có vẻ không đồng đều, đặc biệt là ở các vùng mắt và má, và trán có vẻ rộng và cao. [1]
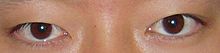
Do trán bất thường, có ít không gian cho các đặc điểm khuôn mặt bình thường phát triển. Điều này dẫn đến hốc mắt nông và xương gò má phẳng. Hốc mắt nông làm cho mắt nổi bật hơn hoặc lồi hơn và khiến mắt bị tách ra nhiều hơn bình thường (hypert Bachelorism). Hốc mắt kém phát triển, xương gò má và hàm dưới khiến khuôn mặt có vẻ phẳng. Hơn nữa, mắt xếch xuống nhỏ cùng với mí mắt (ptosis) làm tăng thêm sự không đồng đều của khuôn mặt. [1]
Di truyền học [ chỉnh sửa ]
SCS xảy ra khi có một đột biến trên nhiễm sắc thể 7 trong khu vực 7p21
SCS thường được di truyền như một tính trạng trội của nhiễm sắc thể. Tuy nhiên, đôi khi, trẻ em có microdeletion 7p21 (nhiễm sắc thể chứa locus chịu trách nhiệm về SCS) phát triển những bất thường mới và thường cho thấy những bất thường đáng kể về thần kinh. Tuổi cha mẹ tăng lên có thể đóng một vai trò trong sự phát triển của các đột biến và bất thường mới. [2]
Phân tích liên kết và sắp xếp lại nhiễm sắc thể cho thấy nguyên nhân của SCS là đột biến gen TWIST (gen nhân tố phiên mã xoắn) nằm trên nhiễm sắc thể 7p21. Gen TWIST mã hóa một yếu tố phiên mã helix-loop-helix (b-HLH) cơ bản kiểm soát sự phát triển trung mô của đầu khi hình thành ống sọ. Hơn 35 đột biến TWIST khác nhau liên quan đến miền b-HLH của protein đã được xác định ở những người bị SCS. Các đột biến bao gồm các đột biến xóa / chèn khung hình, vô nghĩa và xóa khung hình có thể rút ngắn hoặc phá vỡ miền b-HLH. Hầu hết các cá nhân mắc SCS đều bị xóa một vùng lớn trong vùng 7p21, trong đó có vùng mã hóa gen TWIST. [5]
Khi tìm kiếm gen chịu trách nhiệm về SCS, các nhà khoa học tại Trung tâm trẻ em Johns Hopkins bắt đầu nghiên cứu gen TWIST vì nó tác dụng lên chuột. Gen TWIST ở chuột, có chức năng phát triển cơ và xương của mặt, đầu, tay và chân. Những con chuột thiếu cả hai bản sao của gen TWIST đã bị hủy bỏ một cách tự nhiên trước khi sinh và bị dị tật nghiêm trọng bao gồm các chi và dị tật đầu bất thường và sự thất bại của ống thần kinh. Tuy nhiên, những con chuột với một bản sao của gen TWIST không hoạt động đã sống sót. Kiểm tra thêm cho thấy những con chuột này chỉ có khuyết tật nhỏ ở sọ, tay và chân tương tự như những gì nhìn thấy trong SCS. Gen TWIST của chuột nằm trên nhiễm sắc thể 12 ở chuột, tương ứng với nhánh ngắn của nhiễm sắc thể 7 ở người. Với thông tin này, các nhà khoa học bắt đầu phân lập và lập bản đồ gen TWIST của con người trên nhánh ngắn của nhiễm sắc thể người 7. Họ tiết lộ rằng gen TWIST của con người nằm trong cùng khu vực vắng mặt ở những người mắc SCS. Trong khi tìm kiếm các đột biến khác nhau trong gen TWIST ở người, năm loại đột biến khác nhau đã được phát hiện ở những người mắc SCS. Vì không có đột biến nào được nhìn thấy ở những người bình thường không mắc SCS, điều này cung cấp đủ bằng chứng để kết luận rằng gen TWIST là tác nhân gây bệnh của SCS1. Các nhà nghiên cứu cũng đã nghiên cứu gen TWIST trong Drosophila (ruồi giấm) để xác định chức năng của nó. Họ phát hiện ra rằng với sự có mặt của hai phân tử protein TWIST kết hợp với nhau, gen TWIST hoạt động như một yếu tố phiên mã DNA, nghĩa là nó liên kết với chuỗi xoắn kép DNA tại các vị trí cụ thể để kiểm soát gen nào được "bật" hoặc kích hoạt. Phần lớn các đột biến được xác định trong gen TWIST can thiệp vào cách thức protein gắn vào DNA, ngăn chặn sự kích hoạt của các gen khác thường được bật trong quá trình phát triển của thai nhi. [1]
Chẩn đoán [ chỉnh sửa ]
Chẩn đoán trước sinh [ chỉnh sửa ]
Chẩn đoán trước sinh của Hội chứng Saethre-Chotzen trong thai kỳ có nguy cơ cao là có thể thực hiện được, nhưng rất hiếm gặp và hiếm khi được thực hiện. Hơn nữa, điều này chỉ có thể nếu đột biến gây bệnh đã được xác định trong bộ gen của gia đình. Có một vài kỹ thuật khác nhau trong đó xét nghiệm tiền sản có thể được thực hiện. Xét nghiệm tiền sản thường được thực hiện vào khoảng 15 Tuần18, sử dụng nước ối để trích xuất DNA từ các tế bào của thai nhi. Thử nghiệm tiền sản cũng có thể được thực hiện trong tuần 10112 bằng cách sử dụng phương pháp lấy mẫu lông nhung màng đệm (CVS) để lấy DNA từ thai nhi. [6] Gần đây, đã có sự quan tâm tăng lên trong việc sử dụng thiết bị siêu âm để phát hiện bất thường hộp sọ của thai nhi do chưa trưởng thành. Sự hợp nhất của các chỉ khâu sọ. [3]
Chẩn đoán lâm sàng [ chỉnh sửa ]
Chẩn đoán tổng thể về SCS chủ yếu dựa trên các phát hiện lâm sàng và quan sát dựa trên kiểm tra dị hình (đánh giá các khiếm khuyết cấu trúc) đánh giá (X-quang, MRI và CT scan). [5] Chẩn đoán lâm sàng của SCS thường dựa trên sự hiện diện của các đặc điểm sau: [6]
Craniosynostosis
Thông thường nhất là kết quả của sự hợp nhất sớm của chỉ khâu vành, mặc dù bất kỳ chỉ khâu nào khác cũng có thể dẫn đến điều này. [6]
Craniosynostosis thường đi kèm với hình dạng hộp sọ kỳ lạ (ví dụ, brachycephaly [short & broad] ]). [6]
Khi xác định liệu một cá nhân có SCS hay không, bác sĩ sẽ kiểm tra hộp sọ của bệnh nhân và sẽ có thể biết liệu hợp hạch sớm có xảy ra hay không dựa trên hình dạng hộp sọ. [6]
Đường chân tóc thấp, không đối xứng, ptosis (mí mắt rũ xuống) và lác (mắt lác / lười) [6]
Đôi tai nhỏ có âm rõ, phồng lên [6]
Những bất thường ở chân bao gồm búi tóc (hallux valgus), ngón tay ngắn, một phần có màng hình tam giác ngón chân) và phalanx trùng lặp của hallux [6]
Chẩn đoán phân tử / di truyền [ chỉnh sửa ]
Chẩn đoán lâm sàng về SCS có thể được xác minh bằng cách kiểm tra gen TWIST1 (chỉ gen đột biến được biết n để gây ra SCS) cho các đột biến sử dụng phân tích DNA, chẳng hạn như phân tích trình tự, phân tích xóa / sao chép và phân tích tế bào học / phân tích FISH. Phân tích trình tự của exon 1 (vùng mã hóa TWIST1) cung cấp một phương pháp tốt để phát hiện tần số đột biến trong gen TWIST1. Những đột biến này bao gồm vô nghĩa, tên lửa, đột biến trang web mối nối và xóa / chèn vào nội tâm. Phân tích xóa / sao chép xác định các đột biến trong gen TWIST1 không dễ dàng phát hiện bằng phân tích trình tự. Các phương pháp phổ biến bao gồm PCR, khuếch đại đầu dò phụ thuộc vào hệ thống ghép kênh (MLPA) và microarray nhiễm sắc thể (CMA). Phân tích Cytogenetic / FISH gắn nhãn huỳnh quang đánh dấu DNA vào nhiễm sắc thể bị biến tính và sau đó được kiểm tra dưới ánh sáng huỳnh quang, cho thấy các đột biến gây ra bởi sự dịch mã hoặc đảo ngược liên quan đến 7p21. Đôi khi, những người mắc SCS có sự chuyển vị nhiễm sắc thể, đảo ngược hoặc nhiễm sắc thể vòng 7 liên quan đến 7p21 dẫn đến kết quả không điển hình, chẳng hạn như tăng chậm phát triển. [6] Những người bị SCS, thường có chức năng não bình thường và hiếm khi bị suy yếu. Vì lý do này, nếu một cá nhân bị cả SCS và chậm phát triển trí tuệ, thì họ nên kiểm tra gen TWIST1 cẩn thận hơn vì đây không phải là một đặc điểm bình thường của SCS. [1] Xét nghiệm tế bào học và xét nghiệm gen trực tiếp cũng có thể được sử dụng để nghiên cứu gen / khiếm khuyết nhiễm sắc thể. Xét nghiệm tế bào học là nghiên cứu về nhiễm sắc thể để phát hiện sự tăng hoặc giảm của nhiễm sắc thể hoặc đoạn nhiễm sắc thể bằng cách sử dụng huỳnh quang trong lai tạo tại chỗ (FISH) và / hoặc lai genom so sánh (CGH). Xét nghiệm gen trực tiếp sử dụng máu, tóc, da, nước ối hoặc các mô khác để tìm các rối loạn di truyền. Xét nghiệm gen trực tiếp có thể xác định liệu một cá nhân có SCS hay không bằng cách kiểm tra máu của cá nhân đó để tìm đột biến gen TWIST1. [6]
Chẩn đoán phân biệt (điều kiện thường bị nhầm lẫn) [ chỉnh sửa ]
Cho đến gần đây , các chuyên gia thường không đồng ý về việc một bệnh nhân mắc SCS, hội chứng Crouzon, bệnh craniosynostosis hay một số bệnh khác vì các triệu chứng có liên quan mật thiết với nhau, họ thực sự không có cách nào phân biệt giữa tất cả chúng. Tuy nhiên, hiện tại chúng tôi đã thử nghiệm gen trực tiếp, cho phép chẩn đoán chính xác hơn vì nó cho phép chúng được phân biệt với nhau dựa trên gen nào bị đột biến trong từng tình trạng. [1] Sau đây là danh sách các tình trạng thường bị nhầm lẫn / chẩn đoán sai đối với SCS, một số triệu chứng của chúng và mỗi gen đột biến chứa:
Bệnh / Tình trạng
Triệu chứng
Gene đột biến
SCS
Đôi mắt rộng, Đường chân tóc thấp, Mắt rũ xuống, Mạng lưới kỹ thuật số, Tai bị biến dạng, Mắt lác và Vết nứt lòng bàn tay dốc xuống
TWIST1
Hội chứng Robinow-Sorauf
Mắt rộng, vách ngăn lệch, xương sọ phẳng, tai bị biến dạng, mắt lác, hàm nhô ra và sao chép của phalanx xa
TWIST1
Hội chứng Muenke
Mắt rộng, đầu to, Mất thính giác, má phẳng và tai thấp
FGFR3
Hội chứng Crouzon
Mắt rộng, đầu ngắn, Mất thính giác, mắt lồi, mũi bị lõm, tai thấp, Strabismus, cằm lồi, và xương cụt và xương đùi ngắn
FGFR2 & FG
Hội chứng Pfeiffer
Mắt rộng, hàm kém phát triển, mũi bị nghẹt, mất thính giác và mắt lồi
FGFR1 & FGFR2
Hội chứng Apert
Mắt rộng, trán nổi bật, sau sọ phẳng, mắt lồi, tai thấp, mặt phẳng hoặc lõm, ngón tay cái ngắn và ngón tay có màng
FGFR2
Synostosis mạch vành đơn phương bị cô lập
Chỉ có dị tật là sự hợp nhất sớm của chỉ khâu; Nếu không được điều trị, có thể dẫn đến sự bất cân xứng trên khuôn mặt giống với SCS
FGFR (bất kỳ)
Hội chứng Baller-Gerold (BGS)
Đầu rộng ngắn, mắt lồi, trán phẳng, Poikiloderma, biến dạng xuyên tâm với số lượng chữ số giảm, ngón tay cái và bán kính kém phát triển và chậm phát triển
Điều trị và quản lý [ chỉnh sửa ]
Các đoạn xương được cắt bỏ trong sự tiến bộ trước siêu âm
Một đứa trẻ đội mũ bảo hiểm sọ mặt sau phẫu thuật.
kết quả từ SCS thường nhẹ và chỉ yêu cầu một thủ tục tiểu phẫu hoặc không có thủ tục nào cả. Một trong những triệu chứng phổ biến của SCS là sự phát triển của ngắn (brachydactyly), ngón tay có màng và ngón chân rộng (syndactyly). Những đặc điểm này không gây ra bất kỳ vấn đề nào đối với chức năng của tay hoặc chân, và do đó, không có quy trình y tế nào được yêu cầu để khắc phục các bất thường, trừ khi bệnh nhân yêu cầu. Việc quấn ngón tay có thể ảnh hưởng đến gốc ngón tay, dẫn đến chậm phát triển bàn tay trong thời thơ ấu, nhưng điều này không gây ra suy giảm chức năng. Đôi khi, các cá nhân với SCS phát triển các ngón chân rộng vì xương ở cuối các ngón chân đang tự nhân đôi. Điều này đặc biệt thấy ở ngón chân cái, nhưng không cần can thiệp phẫu thuật vì nó không ảnh hưởng tiêu cực đến chức năng chung của bàn chân. Những người có những bất thường ở ngón chân này đi lại bình thường và có thể mang giày dép bình thường. [8]
Trong những trường hợp nghiêm trọng hơn, cần phải phẫu thuật thường xuyên và theo dõi lâm sàng trong suốt quá trình phát triển. Một đứa trẻ sinh ra với khớp thần kinh đơn phương không đối xứng phải trải qua phẫu thuật tạo hình sọ trong năm đầu đời để ngăn ngừa tăng áp lực nội sọ và ngăn ngừa sự bất cân xứng tiến triển trên khuôn mặt. Cranioplasty là một thủ tục phẫu thuật để điều chỉnh xương sọ hợp nhất sớm. Phẫu thuật có tác dụng tái tạo và tái định vị xương và chỉ khâu nhằm thúc đẩy sự phát triển bình thường nhất. [5] Phẫu thuật tạo hình là cần thiết để tiếp tục phát triển và rất quan trọng vì hai lý do chính. Trước hết, hộp sọ cần có khả năng thích ứng với bộ não đang phát triển sau khi sinh con, điều này không thể vì hộp sọ không phát triển nhanh như não miễn là chỉ khâu vẫn hợp nhất. Điều này dẫn đến sự gia tăng áp lực xung quanh não và ức chế não phát triển, khiến cá nhân gặp phải những vấn đề nghiêm trọng, và nếu không được điều trị cuối cùng có thể dẫn đến tử vong. Thứ hai, cranioplasty có thể được yêu cầu cho mục đích ngoại hình. [6] Đây là trường hợp đặc biệt ở những người có khớp thần kinh đơn phương không đối xứng, cần phẫu thuật tái tạo khuôn mặt và hộp sọ. Nếu phẫu thuật tạo hình sọ mặt không được thực hiện, đặc biệt là ở những người mắc hội chứng mạch vành đơn phương, thì sự bất cân xứng trên khuôn mặt sẽ ngày càng tồi tệ hơn theo thời gian, đó là lý do tại sao nên phẫu thuật tạo hình sọ mặt càng sớm càng tốt. [8]
Phẫu thuật cũng có thể được yêu cầu ở những người có vấn đề về thị lực. . Các vấn đề về thị lực thường phát sinh do thiếu không gian trong quỹ đạo mắt và hộp sọ do cấu trúc xương bất thường của khuôn mặt. Không gian giảm cũng có thể dẫn đến ống dẫn nước mắt bất thường hoặc mất tích và tổn thương thần kinh. Phẫu thuật tái tạo thường được yêu cầu để tăng không gian sọ, điều chỉnh hẹp ống dẫn nước mắt và / hoặc ptosis chính xác của mí mắt để ngăn ngừa nhược thị (mắt lười). [1]
Phẫu thuật giữa xương cũng có thể được yêu cầu trong thời thơ ấu để điều chỉnh hô hấp vấn đề, malocclusion, và nuốt khó khăn. Một khe hở vòm miệng cũng được điều chỉnh bằng phẫu thuật và có thể liên quan đến việc sử dụng ống thông khí quản. Nếu cần thiết, một cá nhân sẽ trải qua điều trị chỉnh nha và / hoặc điều trị chỉnh nha sau khi phát triển khuôn mặt hoàn tất. [1] Vì mất thính giác thường liên quan đến SCS, nên kiểm tra thính lực học trong suốt thời thơ ấu. [5]
Sau phẫu thuật tái tạo sọ. một đứa trẻ có thể được yêu cầu đội mũ bảo hiểm đúc hoặc một số hình thức bảo vệ đầu khác cho đến khi xương sọ được đặt đúng vị trí. Điều này thường mất khoảng ba tháng và tùy thuộc vào độ tuổi của trẻ và mức độ nghiêm trọng của tình trạng. Sau khi phục hồi, những người bị SCS trông và hành động hoàn toàn bình thường, do đó, không ai có thể nói rằng họ có SCS. [9]
Dịch tễ học [ chỉnh sửa ]
SCS là phổ biến nhất Hội chứng craniosynostosis và ảnh hưởng đến 1 trên 25.000 đến 50.000 cá nhân. [10] Nó xảy ra ở tất cả các nhóm chủng tộc và dân tộc, và ảnh hưởng đến nam và nữ như nhau. [1] Nếu cha mẹ mang một bản sao của đột biến gen SCS, thì có một 50% cơ hội con của họ cũng sẽ mang một bản sao của đột biến gen, trong trường hợp đó, đứa trẻ có thể hoặc không thể có dấu hiệu của SCS. Cũng có 50% khả năng con của họ sẽ có hai bản sao gen hoạt động, và do đó, sẽ không có SCS. Nếu cả hai cha mẹ đều mang một bản sao của đột biến gen SCS, thì có 25% khả năng con họ sẽ có hai bản sao đột biến gen (vì vậy đứa trẻ sẽ phát triển SCS nghiêm trọng), 25% khả năng con của chúng có hai bản sao bình thường của gen (vì vậy sẽ hoàn toàn bình thường) và 50% khả năng con của họ sẽ mang một bản sao đột biến gen và 1 bản sao bình thường (vì vậy trẻ có thể hoặc không thể hiển thị SCS). [1] Trong những tình huống hiếm hoi, hai cha mẹ bình thường có thể có một đứa trẻ bị SCS do đột biến de novo . Nguyên nhân chính xác của đột biến de novo vẫn chưa được biết, nhưng dường như nó không liên quan đến bất cứ điều gì mà cha mẹ đã làm hoặc không làm trong khi mang thai. [11] SCS do de novo đột biến rất hiếm đến nỗi tỷ lệ của các trường hợp trong quá khứ là không xác định. [6]
Lịch sử [ chỉnh sửa ]
Năm 1931, Haakon Saethre, một bác sĩ tâm thần người Na Uy, đã mô tả tương tự đặc điểm giữa một người mẹ và hai cô con gái. Tất cả chúng đều có các đặc điểm trên khuôn mặt dài và không đều, đường chân tóc thấp, ngón tay ngắn và có màng giữa ngón thứ hai và thứ ba và giữa các ngón thứ hai, thứ ba và thứ tư. Một năm sau, vào năm 1932, F. Chotzen, một bác sĩ tâm thần người Đức, đã mô tả một người cha và hai đứa con trai của ông có những đặc điểm rất giống với người mẹ và con gái của bà, cũng như mất thính giác, tầm vóc ngắn và chậm phát triển tâm thần. Do đó, cái tên Hội chứng Saethre-Chotzen được bắt nguồn từ hai nhà khoa học, những người đã mô tả riêng về tình trạng này mà không có bất kỳ kiến thức nào trước đây về cái kia. [1]
Acrocephalosyndactyly III [12]
Acrocephalosyndactyly type III
Tài liệu tham khảo [ chỉnh sửa ]
^ a b d e f h i j [194545973] k l m n 19659133] o p Blanchford, Stacey L (2 002). Bách khoa toàn thư Gale về rối loạn di truyền . Michigan: Nhóm Gale. trang 1019 bóng1021. SỐ TIẾNG VIỆT87876140.
^ a b c [194545973] [19459] e f g ] h i Allanson, Judith, Cassidy, Suzanne (2010). Quản lý các hội chứng di truyền . New Jersey: John Wiley & Sons, Inc. Trang 230 Tiếng235. ISBN YAM470191415.
^ a b c [194545973] e f g ] h Wynbrandt, James (2008). Rối loạn di truyền và dị tật bẩm sinh . New York: Thông tin về File, Inc. p. 340. ISBN YAM81813939.
^ a b c "Saethre Viện sọ não quốc tế . Truy xuất ngày 28 tháng 10, 2012 .
^ a b d e f [194545973] g h i j 19659133] k l m Clauser L, Galie M. "Hội chứng Saethre-Chotzen" . Mồ côi . Truy cập ngày 28 tháng 10, 2012 .
^ a b d e f [194545973] g h i j 19659133] k l m n , Castyham M. "Hội chứng Saethre Chotzen". NCBI . Truy xuất 28 tháng 10, 2012 .
^ "Hội chứng Saethre-Chotzen". Bệnh viện và Trung tâm y tế trẻ em . Truy cập ngày 25 tháng 10, 2012 .
^ a b Anderson, Peter. "Tiêu đề hỗ trợ sọ não" (PDF) . Được lưu trữ từ bản gốc (PDF) vào ngày 30 tháng 6 năm 2012 . Truy xuất ngày 27 tháng 11, 2012 .
^ "Các lựa chọn phẫu thuật cho bệnh Craniosynostosis". Y học Johns Hopkins . Truy cập ngày 28 tháng 11, 2012 .
^ "Hội chứng Saethre-Chotzen". Bệnh viện nhi Boston . Truy xuất ngày 28 tháng 11, 2012 .
^ "Hội chứng Saethre-Chotzen". Bệnh viện nhi Seattle . Truy cập ngày 28 tháng 11, 2012 .
^ "Sức khỏe của trẻ em: Hội chứng Saethre Chotzen". WebMD . Truy cập ngày 28 tháng 11, 2012 .